About this pathway
Introduction
Valproic acid (VPA) is a branched short-chain fatty acid derived from naturally occurring valeric acid. VPA is used in the treatment of epilepsy and seizures but also migraine, bipolar, mood, anxiety and psychiatric disorders [Article:20798865]. Recent work has explored its use as an adjuvant agent in cancer, HIV therapy, and neurodegenerative disease because of its action as histone deacetylase (HDAC) inhibitor [Articles:19754465, 20132536].
VPA is available in oral, rectal and injectable dosage forms. It is widely used in pediatric epilepsy because of its multiple mechanisms of action and acceptable safety profile [Article:10319910]. The dose requirements for VPA are highly variable (10 fold differences in mean dose in adults) [Article:10594867] and interactions with other drugs are common (discussed below). Therapeutic drug monitoring is commonly used although both the clinical and toxic effects of the drug are considered poorly correlated with total serum concentrations [Article:10319910].
The drug label carries a black box warning for life-threatening adverse drug reactions (ADR) including hepatotoxicity [Articles:21038416, 21544075], teratogenicity [Article:21521026] and pancreatitis [Article:15526953]. Compared to adults, children appear to be at increased risk for severe hepatotoxic reactions to VPA. A series of three retrospective studies of VPA-associated hepatotoxicity in the United States delineated patient age less than two years, polytherapy with enzyme-inducing antiepileptic drugs (AEDs), developmental delay and coincident metabolic disorders as important risk factors for developing this adverse event [Article:3102998]. Although VPA hepatotoxicity may occur at any age, the risk of fatal hepatotoxicity is highest (approximately 1:600) in children less than two years of age receiving concurrent anticonvulsant therapy. Hyperammonemia is also a documented ADR of VPA treatment although this is usually successfully resolved by cessation of VPA and treatment with carnitine [Articles:20456087, 21147182]. The Food and Drug Administration has issued recommendations for patient testing and advises that VPA is contraindicated in patients with known urea cycle disorders (see Clinical PGx tab). However, specific genetic tests for the determination of unsuitable patients are not mentioned.
Pharmacodynamics
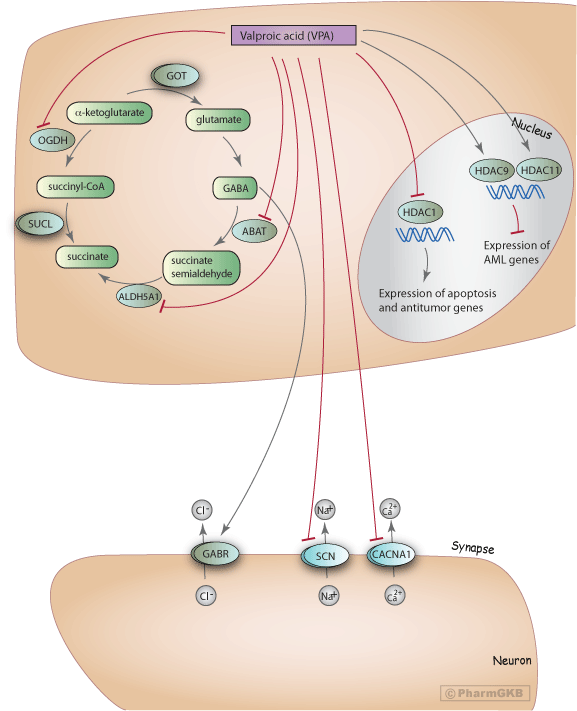
VPA exhibits its pharmacodynamic effects in different ways: it acts on Gamma Amino Butyrate (GABA) levels in the brain, blocks voltage gated ion channels and also acts as HDAC inhibitor (HDACi).
Impairment of the GABAergic inhibitory activity can lead to convulsions, making the control of this pathway a target for anticonvulsants and antiepileptic drugs. GABA is formed from alpha-ketoglutarate through the TCA cycle and metabolized to succinate semialdehyde (SSA) by GABA transaminase (ABAT) and then to succinate by succinate semialdehyde dehydrogenase (ALDH5A1). alpha-ketoglutarate can also be converted to succinyl-CoA through the action of alpha-ketoglutarate dehydrogenase (OGDH), shunting away from the formation of GABA. Ex vivo and in vitro studies have shown that VPA inhibits ABAT and ALDH5A1, both of which are involved in the degradation pathway of GABA [Article:12847559]. One in vitro study also showed that OGDH was inhibited by VPA at high concentration [Article:12847559].
Besides the elevation of the GABA levels, VPA is may also have anti-epileptic activity by reducing the high-frequency firing of neurons by blocking voltage-gated sodium, potassium and calcium channels (including those coded for by CACNA1C, CACNA1D, CACNA1N, and CACNA1F and the SCN gene family)[Articles:8387930, 12847559]. However, whether VPA increases or decreases the conductance of potassium channels is still controversial [Articles:3084227, 12847559, 20798865].
Recently, VPA was demonstrated to be an inhibitor for histone deacetylase 1 (HDAC1) as well as other HDACs [Articles:21508940, 11473107, 11742974], which potentially increases the expression of genes involved in apoptosis and anti-tumor action. Therefore, VPA has been proposed to be a potential anti-tumor agent. In a phase II trial, combination therapy with 5-AZA and VPA was shown to be effective and safe in myelodysplastic syndrome MDS patients with poor prognosis [Article:19638460]. VPA is an activator of HDAC 9 and 11 in cancer cell lines. HDAC inhibitor (like VPA) induced activation/overexpression of specific deacetylases in tumor cells can increase the effectiveness of therapies by promoting selective killing of tumor cells [Article:16121216].
Drug-drug interactions
Over 600 drugs have reported interactions with VPA [Article:20798865]. Several drugs significantly influence the clearance of VPA including aspirin, caffeine, and various antibiotics. Aspirin and caffeine act by displacing VPA binding to serum proteins, inhibiting its clearance [Article:1970771]. Though this may be used to reduce the daily dose of VPA, it can also increase the risk of toxicity of VPA [Article:8549027]. Carbapenem antibiotics increase the glucuronidation of VPA and increase clearance [Article:20798865]. The risk for hepatotoxicity with VPA is greatest in infants under 2 years old treated with additional drugs [Article:20798865]. Drugs that activate cytochrome P450 enzymes increase the clearance of VPA, increase the amount of N-acetylcysteine conjugates of VPA and may have the greatest risk for hepatotoxicity [Article:12614387]. Metabolic studies in children show VPA perturbs organic acid profiles in children, reflecting effects on endogenous pathways corresponding to branched-chain amino acid metabolism and oxidative stress [Article:21544075]. Understanding the molecular pathways underlying these effects may aid in identifying those at greatest risk for ADRs.
VPA can also influence the clearance and metabolism of other drugs. A significant interaction can occur with coadministration of lamotrigine, where VPA doubles its half- life and increases risk of the ADRs Stevens-Johnson syndrome and toxic epidermal necrolysis [Articles:8119045, 8885114, 9421101, 17502552, 19153164]. Interaction with topiramate, another AED, has been shown to increase risk for hyperammonemia and encephalopathy [Articles:15079854, 19952878]. VPA also interferes with HIV medications zidovudine lopinavir and ritonavir [Articles:14986271, 16368918]. A joint panel of the American Academy of Neurology (AAN) and the International League Against Epilepsy (ILAE) guideline suggests "patients receiving valproic acid may require a zidovudine dosage reduction to maintain unchanged serum zidovudine concentrations" [Article:22221159].
Pharmacogenomics
There have been relatively few pharmacogenomic studies of VPA compared to other AEDs carbamazepine and phenytoin. Most have concentrated on well-known polymorphisms in the UGT and CYP candidate genes.
UGT variants
A study of recombinant UGT1A6 proteins showed glucuronidation of VPA was higher for the *2 haplotype (which comprises rs6759892 T>G (Ser7Ala), rs2070959 A>G (Thr181Ala) and rs1105879 A>C (Arg184Ser) compared to the *1 haplotype [Article:15761113]. However, further work in this series with serotonin, another substrate of UGT1A6, found that the results with recombinant *2 did not correlate well with those seen with human liver microsomes genotyped as *2*2.
Recent study on epileptic patients under maintenance with VPA monotherapy and stable seizure control demonstrated that carriers of the variant UGT1A6 19T>G (rs6759892) 541A>G (rs2070959 ) and 552A>C (rs1105879) allele required higher VPA dosages and lower log transformed (concentration-to-dose ratios [CDRs]) than noncarriers. This may be suggestive of higher activities of the UGT1A6 enzymes in patients with the variant genotypes and may need higher VPA maintenance dosages than those without variants [Article:21806385].
CYP variants
In a study of Asian patients with epilepsy, individuals with CYP2A6*4 alleles (which results in almost complete deletion of the CYP2A6 gene) had higher mean plasma VPA concentrations compared to those without [Article:20089352]. In addition those homozygous for CYP2B6*6 (which have severely decreased expression and function) had higher mean plasma VPA concentrations compared to those without this haplotype [Article:20089352]. Carriers of CYP2C9*3 (a reduced activity allele defined by the rs1057910 variant) also showed higher mean plasma VPA concentrations compared to non-carriers [Article:20089352]. These associations were still seen after adjusting for dose, although dose information for the different genotypes was not shown.
In vitro studies of recombinant CYP2C9*2 and CYP2C9*3 proteins and human liver microsomes showed reduced formation of 4-ene-VPA, 4-OH-VPA, and 5-OH-VPA metabolites of VPA [Article:14597963]. However, a study of CYP2C9 variants on production of 4-ene-VPA in vivo showed no significant effects [Article:20602621].
In a small study of Italian patients with myelodysplastic syndromes receiving 5-azacytidine, carriers of the CYP2C19*2 genotype required higher doses of adjuvant VPA to achieve the target VPA plasma concentrations [Article:19638460]. However, the differences between dose ranges were small even though significant (600-1,400mg versus 600-1,000mg; p = 0.0021) and may have been skewed by a few high doses individuals. Also they did not state how many patients were CYP2C19*2 carriers.
In vitro studies of recombinant CYP2C19 protein have not shown production of significant amounts of VPA metabolites (4-ene-VPA, 4-OH-VPA, 3-OH-VPA or 5-OH-VPA)[Article:16945988] making it unlikely that the CYP2C19*2 genotype has a direct result on drug metabolism. A recent study of linkage across the CYP2C gene family in a European population showed complete linkage disequilibrium between CYP2C19*2 and the two CYP2C9 alleles CYP2C9*2 and CYP2C9*3 (PMID: 20665013). This suggests that the association observed for CYP2C19*2 might be due to linked CYP2C9 variants although further studies are needed to clarify the roles of CYP2C9 and CYP2C19 variants and VPA dose and plasma drug levels especially in the context of other drugs.
Additional variants
In a study of symptom prevention in bipolar disorder, rs2269577 was associated with increased likelihood treatment response to VPA [Article:19564049]. This SNP is in the XBP1 gene, which codes for a transcription factor and has been implicated in the pathology of bipolar disorder.
A variant in carbamoylphosphate synthetase I (CPS1), rs1047891, was associated with increased risk of hyperammonemia when receiving combined treatment of VPA with two or more other antiepileptics in people with epilepsy [Article:20456087]. The CPS1 encodes a mitochondrial enzyme that catalyzes the conversion of ammonia to urea in the liver. CPS1 gene expression can be altered by epigenetic mechanisms [Article:21281797] therefore it is conceivable that VPA may influence CPS1 transcription and thus ammonia metabolism. VPA has also been shown to another gene in the urea cycle. The metabolite valproyl-CoA inhibits N-acetylglutamate synthase (NAGS) in rat liver mitochondria [Article:21147182].
In epileptic patients under maintenance with VPA monotherapy a variant in Glutamate (NMDA) receptor subunit epsilon-2 receptor (GRIN2B), rs1019385 (-200T>G) was associated with lower VPA dosages and higher CDRs. Authors found that if the patients are receiving an identical dosage of drug, the patient with the higher CDR will present with a higher plasma drug concentration. Hence, patients with variant genotypes of GRIN2B -200T>G may need lower doses of VPA to achieve the same target therapeutic levels than patients with the TT genotype [Article:21806385].
Rare variants in mitochondrial polymerase gamma gene POLG that result in various disorders including the neurometabolic disorder Alpers-Huttenlocher syndrome, are associated with increased risk for hepatotoxicity with VPA [Articles:21038416, 20138553].
Conclusions
While there is considerable evidence to link the candidate genes for both the pharmacokinetics and pharmacodynamics of valproic acid few studies have identified genomic variants that influence treatment outcome. Many drug-drug interactions have been reported for VPA and this list will likely increase as VPA is used in additional indications where polytherapy is common such as HIV and cancer. A better understanding of VPA pathways should aid in predicting novel drug-drug interactions and avoiding ADRs from concomitant treatments. These may also aid in the design of large genomic studies that validate the effects of the genomic variants already reported and find new influential variants. Complementary metabolomics studies may also aid in profiling patients at risk for ADRs. Though the FDA recommends patient testing on the VPA drug label to avoid prescribing the drug to individuals with urea cycle disorders, the information is lacking about what type of genetic testing and how it should be carried out. While severe urea cycle disorders are rare, there is preliminary evidence that other common variants may also influence risk for ADRs. More work is needed in larger cohorts to examine the role of other candidate gene variants in VPA pharmacokinetics and pharmacodynamics pathways. Finally newer studies correlating genotype phenotype associations with clinical response will be helpful to increase drug efficacy and to reduce drug related toxicity.
Edit history (2)
- 2012-08-06 Create
- 2025-04-10 Update fixed typos