About this pathway
Introduction
Ondansetron is a first-generation serotonin (5-hydroxytryptamine or 5-HT) receptor antagonist which binds to the 5-HT3 receptor. It is a carbazole compound with carbon and nitrogen rings that give it a structural similarity to serotonin, allowing it to bind to the 5-HT3 receptor and exert its clinical effect [Articles:12608887, 27988869].
Ondansetron can be used either alone or in combination with other antiemetics to reduce nausea and vomiting in patients who have undergone surgery or chemotherapy [Articles:1689807, 2146401, 15190136, 27988869]. It has a similar efficacy as the other 5-HT3 receptor antagonists with the exception of granisetron, which has been shown to have a higher efficacy in some studies [Articles:9506240, 11147601, 11760147, 17530482, 17205281]. The most common side effects caused by ondansetron are headache, dizziness and diarrhea, in addition to reports of cardiac effects as a result of ondansetron administration [Articles:12401905, 17530482].
Pharmacokinetics
Ondansetron is generally available as an oral tablet [Articles:1356742, 8129159, 11736884, 15976206]. Other administration forms, including injectable formulations, have been investigated but, at the time of writing, are not being marketed [Articles:17822864, 20020823, 22137964, 24486335, 25581856, 24919508, 26316701]. Oral ondansetron has a Tmax of around 3 hours and a plasma half-life between 3-6 hours [Articles:2533895, 8129159, 15098801, 25581856]. Strikingly, there is no correlation between plasma concentrations of ondansetron and the level of efficacy. Pharmacodynamic factors, rather than pharmacokinetic factors, are considered to be more likely to affect efficacy, as discussed in the Pharmacodynamics section below. [Articles:2533895, 2145930, 15168080, 19967488, 21073811].
The clearance of ondansetron decreases with age in both pediatric and adult subjects [Articles:2533895, 1839313, 1531044, 7554705]. Elderly subjects have an increased Cmax and AUC of ondansetron compared to younger subjects, possibly due to an age-related reduction in hepatic function. Elderly subjects also had an increased volume of distribution at steady-state dosing and an increased elimination half-life [Articles:2533895, 1839313, 1531044]. The exception to this general trend is in subjects under 6 months of age, who have a decreased clearance of ondansetron compared to older pediatric subjects [Articles:19798490, 25670522]. This is likely due to the fact that CYP3A4, which is considered to mediate the majority of ondansetron metabolism, has reduced activity in infants under 6 months old [Articles:8591723, 16802850].
Plasma clearance of ondansetron is significantly reduced in patients with hepatic impairment, leading to an increased AUC and half-life in these patients compared to healthy controls [Articles:8485026, 8690814]. Studies of the pharmacokinetics of oral forms of ondansetron have suggested that there are possible sex-based differences in ondansetron metabolism. Women were found to have a significantly increased AUC and bioavailability of ondansetron in addition to a non-significant increase in Cmax compared to men. [Articles:1531044, 15098801]
The pharmacokinetics of ondansetron during pregnancy have been investigated. In a case study of one pregnant subject, oral clearance of ondansetron increased over the course of gestation, causing a decrease in both the Cmax and the AUC0-8 [Article:27374186]. Ondansetron has also been found to cross the placenta during the early stage of pregnancy and can be found in fetal tissue and amniotic fluid. However, it should be noted that ondansetron is not considered to cause fetal abnormalities [Articles:16584287, 25670522].
As ondansetron is commonly prescribed to control chemotherapy-induced nausea and vomiting (CINV), it is important to understand any interactions between ondansetron and chemotherapeutic agents. Although ondansetron pharmacokinetics appear to be unaffected by the presence of chemotherapeutic agents [Article:7929872], there is evidence that ondansetron can alter the pharmacokinetic parameters of chemotherapy. Oncology patients receiving either cisplatin or cyclophosphamide and concomitant ondansetron had decreased AUCs of both chemotherapeutic agents compared to patients who were not taking ondansetron [Articles:9788577, 10435726]. The exact reason for this interaction is not known but may involve CYP3A4 inhibition in the case of cyclophosphamide [Article:9788577].
A number of other drug-drug interactions involving ondansetron have been reported. The neurokinin-1 receptor antagonist aprepitant, which is often given in combination with a 5-HT3 receptor antagonist such as ondansetron to control nausea and vomiting. In a crossover study of 19 patients given both aprepitant and ondansetron or ondansetron alone found that aprepitant can cause a ~15% increase in the AUC on ondansetron when administered concomitantly [Article:12867217]. This seems to be most likely due to aprepitant’s role as a moderate CYP3A4 inhibitor [Article:12891225], however this interaction is not thought to have a resulting clinical effect on patients [Article:20734049].
Conversely, rifampin reduces exposure to both oral and intravenous ondansetron by significantly reducing both the AUC and half-life of ondansetron. It also reduces the bioavailability of ondansetron by 33%. This is thought to be due to induction of CYP3A4 and possibly CYP1A2 by rifampin [Article:10223773].
Absorption and distribution
Oral ondansetron has a bioavailability of around 60% and is rapidly absorbed into the plasma following administration [Articles:2533895, 10223773]. Exposure to ondansetron is increased by approximately 17% when it is administered following food [Article:7965657]. Once absorbed, around 80% of ondansetron is found bound to plasma proteins [Articles:8690814, 22112579], with reduced binding seen in patients with hepatic impairment [Article:8690814].
Metabolism
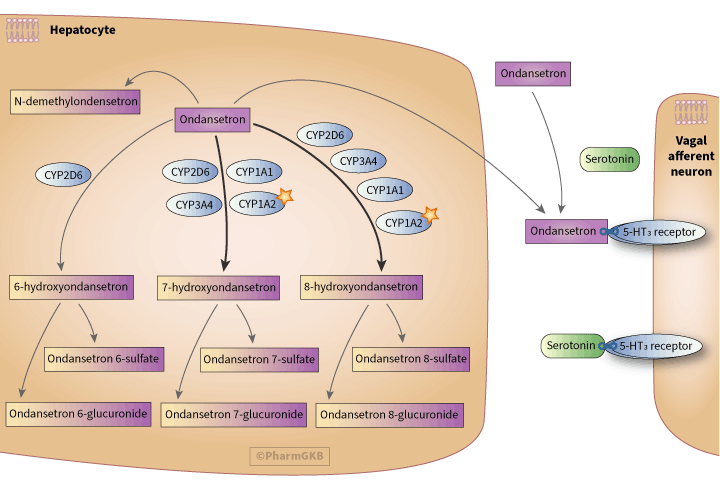
Ondansetron is subject to first-pass metabolism in the liver [Articles:8690814, 16528908]. Following this, it is primarily metabolized by hydroxylation at the 6, 7 or 8 positions of the indole ring [Articles:2533904, 11114092] with some minor metabolites also formed, including N-demethylondansetron [Article:8013282]. These hydroxylated metabolites can be detected in serum but 7-hydroxyondansetron and 8-hydroxyondansetron tend to be the predominant ones [Articles:2533904, 17701832]. These hydroxylated metabolites can then be subsequently glucuronidated or sulfated [Article:2533904].
The metabolism of ondansetron is carried out by multiple enzymes, including CYP1A1, CYP1A2, CYP3A and CYP2D6 [Article:8013282]. Although CYP3A is considered to mediate the majority of ondansetron metabolism [Article:8591723], formation of the metabolites 7-hydroxyondansetron and 8-hydroxyondansetron appears to be mostly carried out by the enzyme CYP1A2, indicated by the star icon on the pathway image [Article:8591723]. CYP2D6 also has a minor role in the hydroxylation of ondansetron, mediating approximately 30% of total ondansetron metabolism [Articles:8013282, 8591723, 8861656].
The different contributions of CYP3A and CYP2D6 to ondansetron metabolism can be explained by the differences in ondansetron’s binding affinity to each enzyme. Ondansetron forms four π-π stacking interactions with CYP3A4, resulting in an increased binding affinity compared to that for CYP2D6, where ondansetron only forms one π-π stacking interaction and a hydrogen bond [Article:20201779].
It should be noted that many in vitro experiments which investigate ondansetron metabolism use concentrations of ondansetron which are higher than the peak plasma concentrations of ondansetron seen in vivo [Articles:8013282, 12416899]. Caution should therefore be used when extrapolating the results of these in vitro experiments to ondansetron metabolism in vivo. Indeed, at least one study has found that data from in vitro metabolism experiments have little correlation with data from in vivo experiments [Article:17701832].
Excretion
Around 10% of the original dose of ondansetron is excreted unchanged in the urine with a further 34-43% excreted in the urine as metabolites within 24 hours of administration [Article:2533895].
Pharmacodynamics
Ondansetron prevents the binding of serotonin released from intestinal enterochromaffin cells to 5-HT3 receptors on adjacent vagal afferent nerves. This blockade of 5-HT3 receptors reduces nausea and vomiting by decreasing vagus nerve signaling and the subsequent release of serotonin in the brainstem [Article:11090957]. It is important to note that serotonin signaling is not the only mechanism by which nausea and vomiting can be stimulated and, as a result, ondansetron cannot be used to treat all cases of nausea and vomiting [Article:11090957].
Ondansetron has been found to occupy around 50% of 5-HT3 receptors, however there are large interindividual difference in receptor occupancy [Article:19967488]. Ondansetron efficacy appears to be at least partially correlated to the level of 5-HT3 receptor occupancy [Articles:15168080, 19967488].
The binding of ondansetron to the 5-HT3 receptor has been studied in some detail. Work using the murine 5-HT3 receptor has shown that binding is mediated by cation-π interactions between ondansetron and the Trp183 residue of the receptor. This is a similar mode of binding as when serotonin binds to the 5-HT3 receptor [Article:22873819]. Unlike palonosetron, another 5-HT3 receptor antagonist, binding of ondansetron does not cause internalization of the 5-HT3 receptor [Article:19836386].
In vitro work using a range of animal and human proteins suggest that, in addition to binding to the 5-HT3 receptor, ondansetron may interact with other receptors and transporters, such as MATE1 [Articles:2164935, 11212100, 16115980, 23241029, 24690261]. However, the relevance of these findings to the actions of ondansetron in humans has yet to be determined.
There is a documented pharmacodynamic interaction between ondansetron and serotonin reuptake inhibitors (SRIs), such as fluoxetine, which can lead to serotonin syndrome, where a patient experiences symptoms such as hyperthermia, agitation and tremor as a result of increased serotonin levels [Article:25038602]. Patients receiving ondansetron to treat CINV and a concomitant SRI were more likely to experience vomiting episodes than patients who had never been treated with an SRI, suggesting that SRIs reduce the efficacy of ondansetron [Articles:8590698, 22644261]. This is thought to be due to increased levels of free serotonin as a result of the action of the SRI, which increases competition between serotonin and ondansetron for binding to serotonin receptors. Conversely, ondansetron has been found to augment the actions of SRIs and could be useful for the treatment of conditions such as obsessive-compulsive disorder in patients who have not responded to SRI treatment alone [Articles:24850229, 24406025, 25697477].
Ondansetron has also been found to affect tramadol requirements in patients, with patients taking concomitant tramadol and ondansetron having decreased analgesia and increased tramadol dose requirements compared to patients taking tramadol only [Articles:11323369, 12032025, 21952250, 25490944]. However, it should be noted that not all studies have replicated this observation [Article:20488759]. It has been postulated that the decrease in tramadol analgesia is due to blockade of 5-HT3 receptors by ondansetron, which prevents the binding of serotonin molecules released by the actions of tramadol [Article:12032025].
Ondansetron is known to prolong the QT interval of patients in a dose-dependent manner [Articles:25439413, 24372925]. Work on the effects of ondansetron on cardiac ion channels suggests that ondansetron is able to interact with the inactive state of the cardiac sodium channel Nav1.5. Nav1.5 is encoded for by the gene SCN5A. This interaction causes the channel to become blocked [Articles:11046096, 27401036]. Ondansetron has also been found to be capable of blocking cardiac potassium channels with an increased affinity for these channels over sodium channels [Article:11046096].
Pharmacogenetics
Variation in CYP2D6 activity can impact on the pharmacokinetics of ondansetron, ultimately affecting its efficacy. CYP2D6 ultrarapid metabolizers (UMs) have decreased exposure and a reduced response to ondansetron, as measured by an increased number of vomiting episodes following treatment compared to CYP2D6 normal metabolizers (NMs) [Articles:12065557, 15731591]. As a result, the Clinical Pharmacogenetics Implementation Consortium (CPIC) have published a clinical guideline covering ondansetron advising that CYP2D6 UMs are prescribed an alternative antiemetic medication, such as dolasetron [Article:28002639].
The response of CYP2D6 intermediate and poor metabolizers (IMs and PMs) to ondansetron is less clear. While some work has indicated that IMs and PMs have an increased exposure to ondansetron [Articles:12065557, 21596874], a notable effect of these phenotypes on a patient’s response has not been documented [Article:12065557]. In addition, other studies have found no effect of a PM or IM phenotype on the efficacy or pharmacokinetic parameters of ondansetron [Articles:8018461, 21840870]. It should be noted that Perwitasari et al. did not identify any CYP2D6 PMs in their cohort.
Detailed analysis of the effect of variation in CYP2D6 and CYP3A5 activity on ondansetron pharmacokinetics has found that genetic variation in CYP2D6 affects exposure to the S-ondansetron enantiomer, but not R-ondansetron [Article:21596874]. By contrast, the AUC of R-ondansetron was increased in patients carrying the non-functional CYP3A5*3 allele [Article:21596874].
The synonymous variant rs1045642 in _ABCB1 may affect ondansetron efficacy with the G allele associated with a reduced response to ondansetron while patients carrying the AA genotype were more likely to be free of vomiting following ondansetron treatment [Articles:16338277, 21840870, 25012726]. One study has expanded on this and found that the CTG haplotype consisting of the variants rs1045642, rs2032582 and rs1128503 is associated with a reduced response to ondansetron [Article:16338277].
Work on the relationship between the transporter SLC22A1 and the efficacy of ondansetron has found that patients who do not have any active SLC22A1 alleles may experience an increased efficacy of the drug [Article:20921968]. However, in vitro work by the same group indicated that SLC22A1 was not involved in ondansetron transport so the nature of the effect of inactive SLC22A1 alleles on ondansetron efficacy remains unknown [Article:20921968]. The conflicting results warrant further investigations to determine the role of SLC22A1 on ondansetron exposure.
A study into the effects of polymorphisms in the HTR3A gene, which encodes the 5-HT3A subunit of the 5-HT3 receptor, found no statistically significant effects of any variants on the efficacy of ondansetron [Article:15115912]. Tremblay et al. found that homozygotes for the del allele of rs45460698 in the promotor of _HTR3B, which encodes the 5-HT3B subunit of the 5-HT3 receptor, had significantly more episodes of vomiting when treated with ondansetron compared to heterozygotes or homozygotes for the AAG allele [Article:12775740]. However, this association lost significance following multiple testing correction at one of the two timepoints assessed. Both Tremblay et al. and Perwitasari et al. noted nonsignificant trends for rs45460698 heterozygotes to have increased nausea and vomiting following ondansetron therapy compared to AAG homozygotes [Articles:12775740, 21840870].
Conclusion
The pharmacokinetics and pharmacodynamics of ondansetron are fairly well characterized, although there is an absence of information about the transport of both drugs. While there is clinical guidance regarding the use of ondansetron in CYP2D6 UMs, further work is warranted to elucidate whether the CYP2D6 PM phenotype has an impact on a patient’s response to either drug and to identify additional variants in other metabolizing enzymes, such as CYP1A2, which may have pharmacogenetic effects.
Reactions & interactions (22)
-
Activation
serotonin → 5-HT3 receptor
-
Biochemical Reaction
6-hydroxyondansetron → ondansetron 6-sulfate
-
Biochemical Reaction
ondansetron → N-demethylondansetron
-
Biochemical Reaction
7-hydroxyondansetron → ondansetron 7-sulfate
-
Biochemical Reaction
8-hydroxyondansetron → ondansetron 8-sulfate
-
Biochemical Reaction
6-hydroxyondansetron → ondansetron 6-glucuronide
-
Biochemical Reaction
ondansetron → 6-hydroxyondansetron
-
Biochemical Reaction
ondansetron → 7-hydroxyondansetron
-
Biochemical Reaction
7-hydroxyondansetron → ondansetron 7-glucuronide
-
Biochemical Reaction
ondansetron → 8-hydroxyondansetron
-
Biochemical Reaction
8-hydroxyondansetron → ondansetron 8-glucuronide
-
Catalysis
CYP2D6 → Biochemical Reaction
-
Catalysis
CYP1A1 → Biochemical Reaction
-
Catalysis
CYP2D6 → Biochemical Reaction
-
Catalysis
CYP3A4 → Biochemical Reaction
-
Catalysis
CYP1A2 → Biochemical Reaction
-
Catalysis
CYP1A2 → Biochemical Reaction
-
Catalysis
CYP1A1 → Biochemical Reaction
-
Catalysis
CYP2D6 → Biochemical Reaction
-
Catalysis
CYP3A4 → Biochemical Reaction
-
Inhibition
ondansetron → 5-HT3 receptor
-
Transport
ondansetron → ondansetron
Edit history (6)
- 2018-07-24 Create
- 2018-11-30 Update Updated pathway image and description to reflect reviewer comments from upcoming publication of this pathway.
- 2019-09-11 Update Added link to publication
- 2024-08-30 Update Removed CYP3A from gpml and replaced with CYP3A4. Updated figure.
- 2025-07-17 Update Redirected link from CYP3A (non-specific) to CYP3A4
- 2025-07-17 Update Removed broken link to guideline annotation