About this pathway
Introduction
Ivacaftor is one of the first drugs developed to treat an underlying cause of cystic fibrosis (CF) rather than the symptoms. It is a selective small molecule potentiator of the Cystic Fibrosis Transmembrane Conductance Regulator protein (CFTR). Potentiators are a new drug class of CFTR modulators designed to restore CFTR function [Article:24656117]. The Food and Drug Administration (FDA) granted ivacaftor approval in 2012 as it is indicated in a subset of patients with particular CFTR variants, specifically class III variants. Approximately 30,000 people have CF in the United States, which is below the 200,000 patient threshold for orphan drugs in the United States [Article:24656117]. Ivacaftor alone is not approved for F508del which is the most common CFTR variant in the USA [Article:23616732]. Additional modulators lumacaftor, elexacaftor and tezacaftor were designed to be effective for F508del.
CF is an autosomal recessive disorder characterized by sweat chloride concentration above 60 mmol/L, leading to progressive obstructive lung disease and premature mortality, as well as problems in the liver, pancreas, vas deferens, and intestine [Article:24534272]. CF affects approximately 70,000 people worldwide, including approximately 1 of every 3500 infants born in the United States [Article:23457166], and is caused by inheriting two detrimental copies of the CFTR gene (Cystic Fibrosis Foundation). The CFTR protein is a chloride channel, which maintains ion and water balance intra- and extra-cellularly [Article:24561283]. The channel opens and closes through ATP binding, hydrolysis, and phosphorylation, which changes the protein conformation to allow chloride ions to flow through the ion gradient between the intracellular and extracellular regions [Article:24534272]. Defective CFTR function prevents the flow of chloride ions across epithelial cells, which leads to the mucus buildup, infection, inflammation, and progressively decreasing lung function that is characteristic of cystic fibrosis [Article:24561283].
Pharmacokinetics
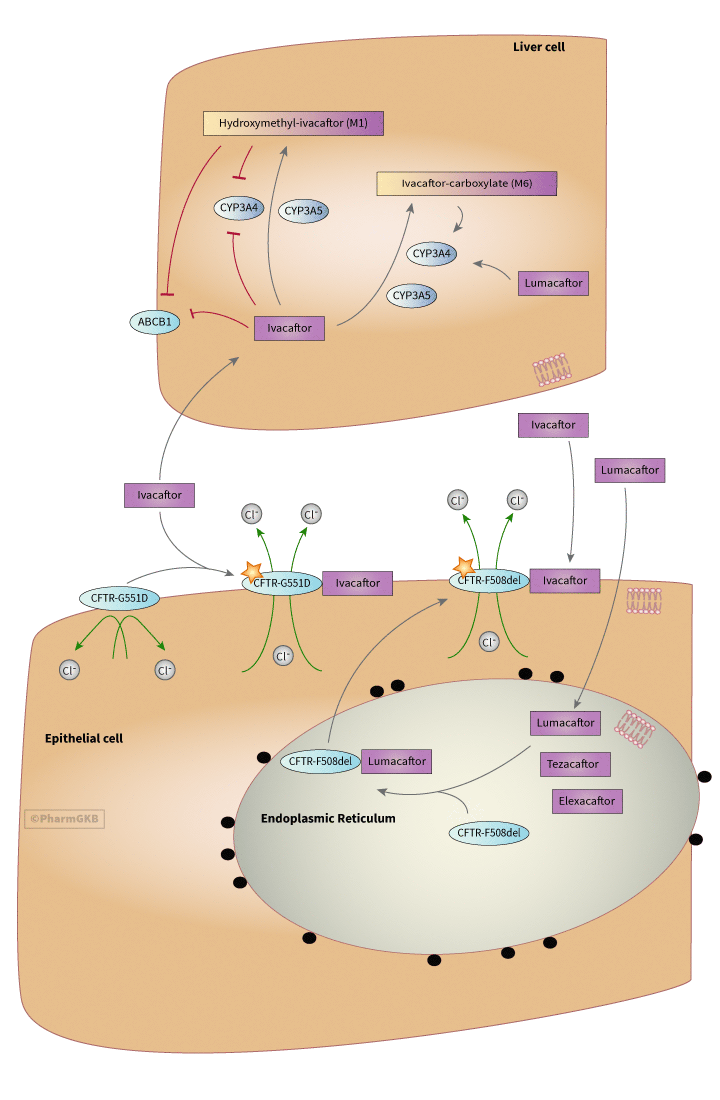
Administered as an oral dose, ivacaftor absorbs readily from the gut, but has low solubility in water (< 0.05 ug/mL) Committee for Medicinal Products for Human Use, Kalydeco: Assessment Report. 2012, European Medicines Agency. Taking a 150 mg dose of ivacaftor with a high fat meal improves absorption, increases AUC by 2.5 times, and delays Tmax from 3 to 5 hours [Article:26968005]. Dose/time pharmacokinetics follows a linear profile to a dose of 250 mg, though Cmax plateaus for doses 375 mg and higher Committee for Medicinal Products for Human Use, Kalydeco: Assessment Report. 2012, European Medicines Agency. Ivacaftor is transported in the plasma highly bound (99%), preferentially to alpha-1-acid glycoprotein but also to albumin, to its site of action, which is the apical membrane of epithelial cells [Articles:26968005, 25148205]. However, no drug-drug interactions related to protein binding competition are expected Committee for Medicinal Products for Human Use, Kalydeco: Assessment Report. 2012, European Medicines Agency. As shown in the figure, ivacaftor is metabolized in the liver by cytochrome P450 3A (CYP3A), including both CYP3A4 and CYP3A5, into a metabolite hydroxymethyl-ivacaftor (M1), which is considered to be active with a potency 1/6th that of ivacaftor itself, and the inactive metabolite ivacaftor-carboxylate (M6), which not considered active and has an activity level 1/50th that of ivacaftor [Article:25103957]. Elimination of the parent drug and metabolites occurs predominantly (87%) through the bile, with 22% being the M1 metabolite and 43% being the M6 metabolite [Article:25148205]. Ivacaftor has a half life of 12-14 hours Committee for Medicinal Products for Human Use, Kalydeco: Assessment Report. 2012, European Medicines Agency.
Ivacaftor and M1 are thought to be weak inhibitors of CYP3A4, increasing midazolam area under the curve of drug concentration in the plasma over time (AUC) by 54% [Article:25103957]. They are also weak inhibitors of P-glycoprotein (ABCB1), increasing digoxin AUC by 32% [Article:25103957]. CYP3A inducers, such as rifampin, carbamazepine and phenytoin, reduce exposure to ivacaftor and CYP3A inhibitors increase exposure to ivacaftor [Articles:25103957, 26581802]. When administered with known CYP3A inhibitors, it is recommended that the frequency of ivacaftor administration be reduced from twice to once daily Committee for Medicinal Products for Human Use, Kalydeco: Assessment Report. 2012, European Medicines Agency.
Pharmacodynamics
Ivacaftor is indicated for use in CF patients who carry at least one of 11 approved genetic variants that affect the gating capacity of CFTR, most of which are class III variants, which affect the activation of the chloride channel of CFTR and thereby inhibit normal chloride movement [Articles:21083385, 12503104]. These variants are listed in the table. The functional consequence of class III variants means that CFTR localizes to the apical cell membrane as normal, but cannot undergo cAMP-mediated activation and so is nonfunctional [Articles:12503104, 26390337]. Ivacaftor is a selective potentiator of CFTR and is believed to stabilize the open state of the channel, enabling chloride transport [Article:23440202]. The exact mechanism of how this potentiation works is unknown, though it may be through decoupling the gating cycle and ATP hydrolysis cycle, or by increasing the ATP-dependent opening rate and slowing the closing rate [Articles:23440202, 24796242]. It is believed that by binding to CFTR in the epithelial cell membrane, ivacaftor improves the function of both CFTR with gating mutations and CFTR with normal function [Articles:25148205, 23440202]. Initial validation studies were conducted in patients with CF and at least one G551D CFTR gating variant (c.1652G>A, rs75527207); treatment was reported to improve lung function by >10%, enough to see a marked improvement in symptoms of CF [Article:22293084]. Ivacaftor treatment was reported to result in improved pulmonary function tests as measured by the Forced Expiratory Volume in 1 second (FEV-1) by 10.4 - 17.5% over the course of 24 weeks, and this difference was maintained to the trial end at 48 weeks [Articles:24461666, 22047557]. Ivacaftor treatment was reported to improve weight gain (3.7 kg on ivacaftor compared with 1.8 kg on placebo over 24 weeks) and to decrease chloride concentration compared to baseline (-55.5 mmol/L ivacaftor compared to -1.8 mmol/L on placebo) in clinical trials with 6-11 year olds [Article:24461666]. Changes in sweat chloride concentrations are often used as a biomarker of drug bioactivity for cystic fibrosis, though there is debate as to the validity of this measure [Articles:23276841, 24258833, 24660233]. Improvement in chloride transport was reported in patients 12 years and older [Article:22047557]. Respiratory improvements were reported after 2 weeks of treatment with ivacaftor compared to placebo [Article:22047557].
Trials in patients homozygous for the F508del variant (c.1521_1523delCTT; rs113993960 or rs199826652), which is not a gating variant and instead prevents CFTR protein from exiting the endoplasmic reticulum and localizing to the cell membrane, showed no improvement with ivacaftor treatment alone [Article:19846789]. However, a combination drug of ivacaftor with lumacaftor (VX-809) was approved by the FDA and European Medicines Agency (EMA) in 2015 for use in patients homozygous for the F508del allele [Article:26417173]. Lumacaftor is reported to restore CFTR function to about 15% of normal in lower airway epithelial cells derived from patients homozygous for the F508del allele by chaperoning protein folding [Article:26581802]. By ameliorating the folding malfunction caused by that allele, lumacaftor is thought to enable the localization of F508del CFTR to the cell membrane [Article:26581802]. When lumacaftor is combined with ivacaftor, the F508del CFTR folding repair from lumacaftor combined with increased potentiation from ivacaftor is reported to result in a 30% improvement of CFTR function in lower airway epithelial cells derived from individuals homozygous for the F508del allele [Article:26581802]. Phase 2 and 3 trials have also shown clinically meaningful efficacy in patients homozygous for the F508del allele, with results measured by change in chloride concentration and FEV-1 [Article:26581802]. Additional modifiers tezacaftor and elexacaftor target the F508del variant [Article:38249954]. Approximately 90% of cystic fibrosis patients are eligible for ivacaftor containing regimens [Article:38249954].
Pharmacogenetics
CFTR has two nucleotide binding domains and two membrane spanning domains, and variants can occur throughout all regions [Article:21652558]. Shuttled from the golgi to the apical membrane in secretory vesicles, CFTR turns over at a rate of 10% per minute and has a half life of 12-24 hours [Article:21652558]. More than 2000 variants have been identified in the CFTR gene, though only 127 have been confirmed to be pathogenic. The American College of Medical Genetics guidelines include 23 of these variants in their recommended panel for determining carrier status for cystic fibrosis [Article:21422883]. Pathogenic variants in CFTR are categorized into 5 classes based on their effect on CFTR function. Class I variants affect biosynthesis of CFTR, producing truncated or unstable protein that is quickly degraded. Class II variants affect protein folding, which can reduce protein stability and appropriate localization. The F508del variant, which is a class II variant and prevents localization to the cell membrane, is identified in over 90% of CF cases [Article:24534272]. Class III variants, those that are targeted by ivacaftor, fold and localize appropriately, but cannot be regulated by ATP or phosphorylation and so do not have gating function. Class IV variants also affect ion flow, but through decreased permeability of chloride ions, and class V variants usually result in splicing changes that reduce the amount of CFTR expressed in the cell membrane [Article:21652558]. The most important genetic variants to consider in ivacaftor treatment are the class III variants in CFTR that cause problems with gating. The G551D variant is the most common gating variant and that allele is thought to be present in about 4-5% of patients with CF [Article:21083385]. The FDA and EMA originally approved the use of ivacaftor in the United States and in Europe in 2012 to treat cystic fibrosis caused by the G551D gating variant [Article:25148205]. Since then, approval has been expanded to include other gating variants. The list of variants approved for treatment with ivacaftor is found in the table and includes G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, and S549R [Articles:25148205, 22293084]. These variants each occur in less than 1% of cystic fibrosis patients [Articles:24561283, 22293084]. In addition to these other class III variants, the FDA has approved ivacaftor for treatment of patients with the R117H variant, a class IV variant that is primarily a conductance variant, but also has defective gating activity [Article:26324139].
Variants in CFTR approved for treatment with ivacaftor
| Amino Acid Change | rs number | cDNA change |
|---|---|---|
| R117H | rs78655421 | 350G>A/C/T |
| G178R | rs80282562 | 532G>A |
| S549R | rs121908755 | 1646G>A/T |
| S549R | rs121908755; rs121909005 | 1645A>C; 1647T>G |
| G551S | rs121909013 | 1651G>A |
| G551D | rs75527207 | 1652G>A |
| G1244E | rs267606723 | 3731G>A/T |
| S1251N | rs74503330 | 3752G>A |
| S1255P | rs121909041 | 3763T>C |
| G1349D | rs193922525 | 4046G>A |
Conclusion
Ivacaftor monotherapy potentiates CFTR protein with defective gating capabilities, the most common cause of which is the G551D variant. Provided CFTR localization is restored with lumacaftor in patients homozygous for the F508del allele, ivacaftor can also improve chloride transport in these individuals. While the specific mechanism of action is unknown, ivacaftor is thought to restore the flow of chloride ions through the cell membrane via CFTR, and thereby reduce the symptoms of CF in patients with a gating defect.
Reactions & interactions (28)
-
Activation
ivacaftor carboxylate → CYP3A4
-
Activation
ivacaftor-CFTR → Transport
-
Activation
CFTR → Ivacaftor-CFTR
-
Activation
elexacaftor → CFTR
-
Activation
CFTR → lumacaftor-CFTR
-
Activation
lumacaftor → CYP3A4
-
Activation
ivacaftor → Ivacaftor-CFTR
-
Activation
tezacaftor → CFTR
-
Activation
lumacaftor → lumacaftor-CFTR
-
Activation
Ivacaftor-CFTR → Transport
-
Activation
CFTR → ivacaftor-CFTR
-
Activation
ivacaftor → ivacaftor-CFTR
-
Biochemical Reaction
ivacaftor → hydroxymethyl ivacaftor
-
Biochemical Reaction
ivacaftor → ivacaftor carboxylate
-
Catalysis
CYP3A4 → Biochemical Reaction
-
Catalysis
CYP3A5 → Biochemical Reaction
-
Catalysis
CYP3A5 → Biochemical Reaction
-
Catalysis
CYP3A4 → Biochemical Reaction
-
Conversion
lumacaftor-CFTR → lumacaftor
-
Conversion
lumacaftor-CFTR → CFTR
-
Inhibition
ivacaftor → ABCB1
-
Inhibition
ivacaftor → CYP3A4
-
Leads To
hydroxymethyl ivacaftor → elimination
-
Leads To
ivacaftor carboxylate → elimination
-
Transport
chloride → chloride
-
Transport
lumacaftor → lumacaftor
-
Transport
CFTR → CFTR
-
Transport
chloride → chloride
Edit history (7)
- 2016-09-19 Create
- 2024-02-23 Update Updates to figure: removal of SLCO1B1, addition of tezacaftor and elexacaftor in PD section, inhibition arrows for M1 and activation arrows for M6 and lumacaftor.
- 2024-02-26 Update Updates to text to fix broken links to EMA assessment document, to add information about tezacaftor and elexacaftor and remove incorrect unsupported sentence on SLCO1B1 and M6.
- 2024-03-14 Update Updates to gpml to remove SLCO1B1, fix problems with format on title and add lumacaftor to PK, elexacaftor and tezacaftor to PD.
- 2024-03-14 Update Added related pathways
- 2024-05-07 Update fixed typo
- 2024-08-30 Update Removal from gpml of inhibition of CYP3A generic to CYP3A4.