About this pathway
Background
Acyclovir (ACV) and ganciclovir (GCV) are acyclic analogs of guanosine that were approved for medical use in 1982 and 1989, respectively. These molecules are commonly prescribed for treating infections caused by herpes simplex viruses (HSV-1 and HSV-2) or varicella-zoster virus (VZV). Additionally, GCV is used as a first-line treatment for cytomegalovirus (CMV) infections (including CMV retinitis and congenital infection) and the oral prodrug valganciclovir (VGCV) is the preferred drug for preventing CMV disease in immunocompromised patients such as transplant recipients, patients positive for human immunodeficiency virus (HIV), and patients with cancer undergoing chemotherapy [Article:8786764]. Large interpatient variability has been described for ACV and GCV that results in differences in toxic effect occurrence and efficacy, with diminished efficacy being characterized by an increase in viral charge after treatment. This variability mainly depends on the characteristics of the drugs and patients and on the clinical indication. Finally, the increasing incidence of viral resistance constitutes an additional challenge for clinicians. Although the drugs are globally well tolerated, some patients may experience severe nephrotoxicity and neurotoxicity after ACV treatment or hematologic and gastrointestinal toxicity after GCV treatment. The evidence for pharmacogenetic interactions has notably implicated variants of the NUDT15 and ABCC4 genes as impairing ACV and GCV efficacy [Articles:34234136, 34192523]. This review describes the metabolism, transport, and mechanism of action of ACV and GCV with a view to decipher the existing interpatient variability, and it supports these drugs being considered as candidates for use in personalized medicine.
Clinical Pharmacokinetics
As structural analogs, ACV and GCV present similar pharmacokinetics profiles. Their poor oral absorption limits their respective bioavailabilities (around 10%–20% for ACV and 8.5% for GCV, with the latter being slightly improved by simultaneous food intake [Articles:6355048, 7585846, 8690817]) and supports their intravenous administration. This limitation was countered by the development of the respective oral pro-drugs valacyclovir (VACV) and valganciclovir (VGCV), which are converted into ACV and GCV by specific intestinal, hepatic, and renal hydrolases [Article:12732646]. These drugs showed substantially improved bioavailabilities of 54.5% and 60%, respectively, when compared to their respective parent drugs [Article:20367273]. ACV is characterized by dose-independent and linear pharmacokinetics after intravenous (I.V.) administration [Article:227639]. Plasma protein binding is low for both molecules (15.4 ± 4.4% for ACV and 1%–2% for GCV), and they can both penetrate the blood–brain barrier [Articles:7102704, 11465876]. Most of a given ACV or GCV dose is eliminated as unchanged drug in the urine through glomerular filtration and active tubular secretion [Articles:7102704, 2847287]. The renal clearances of these drugs correlate strongly with creatinine clearance, and their mean half-lives after I.V. administration are similar, varying between 2.5 and 3 hours for ACV and reaching 4 hours for GCV in adults [Articles:6359082, 2847287]. The high interindividual variability described for ACV and GCV is partially a reflection of renal function and age and justifies reinforced monitoring of biological functions [Articles:19414579, 34596243]. Indeed, renal impairment markedly alters ACV and GCV kinetics by increasing the half-life by up to 10 times for ACV and by 3–20 times for GCV, depending on the degree of renal dysfunction [Articles:7102702, 2847287, 12189361]. Consequently, dose adjustment for the two drugs is based on a patient’s creatinine clearance . The pharmacokinetics of ACV are similar in pediatric patients, with a mean protein-binding rate of 15% (range: 9%–33%) and a primary renal elimination half-life of 2–5 hours for the recommended dosage of 30–45 mg/kg/day in three divided doses that can be increased up to 60 mg/kg in a high-dose regimen [Article:26619466].
Metabolism and transport
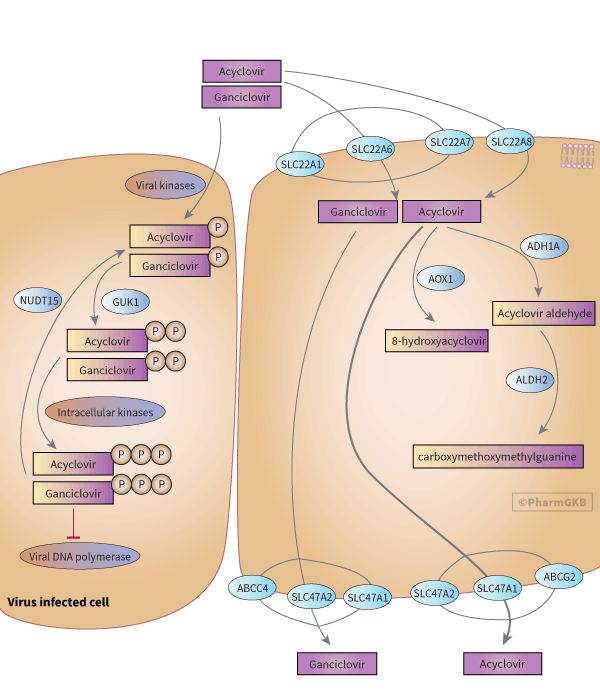
Neither ACV nor GCV is metabolized in the liver or taken in charge by cytochromes P450s. ACV's main metabolite is 9-carboxymethoxymethylguanine (CMMG) (accounting for 8.5% to 14.1% of the ACV dose recovered in urine [Article:7102704]). The mechanism of formation of CMMG has been proposed by Gunness et al. [Article:22005269] after detecting an aldehyde intermediate issued from the oxidation of ACV by the alcohol dehydrogenase 1A (ADH1A) (from class I). The subsequent oxidation of this aldehyde derivative by the aldehyde dehydrogenase 2 (ALDH2) leads to the formation of CMMG. ACV is also transformed, albeit to a much lesser extent, into 8-hydroxyacyclovir (8-OH-ACV, less than 1% of the dose recovered in urine) by the aldehyde oxidase (AOX1) [Articles:7102704, 20038622]. Patients with impaired renal function exhibit a higher urinary excretion rate for metabolites (21%–44% of the dose) [Article:6355048]. Renal elimination of unchanged ACV and GCV in the proximal tubule is mostly supported by uptake transporters from the organic anion transporter (OAT) and organic cation transporter (OCT) families. Most studies of GCV and ACV cell transport have focused on the parental drugs, whose transport was demonstrated to be mediated by OAT1 (encoded by the SLC22A6 gene) and OCT1 (encoded by SLC22A1) [Article:11861798]. In addition, inhibition of OAT1 and OAT3 (encoded by SLC22A8) by benzylpenicillin (an antibiotic that undergoes renal excretion like ACV) was followed by modification of the pharmacokinetics of ACV in animals (GCV was not tested in the study), resulting in an increase in the area under the plasma concentration curve (AUC) and half-life and a decrease in plasma clearance of the drug [Article:23744435]. However, the lack of consistency among the results of different studies precludes the definite implication of OAT1 and OAT3; this inconsistency is highlighted by the in vitro study of Cheng et al. [Article:22190696], who observed ACV and GCV transport only by OAT2 (encoded by SLC22A7) and emphasized the substrate-specificity of the transporters.
Excretion via the urine occurs through the multidrug and toxin extrusion proteins MATE1 (encoded by SLC47A1) and MATE2-K (encoded by SLC47A2), with ACV having a significantly higher substrate affinity for MATE1 [Article:17509534]. The role of ATP-binding cassette transporters in the efflux of ACV and GCV has been mostly demonstrated in vitro. Breast cancer resistance protein (BRCP) (encoded by ABCG2) was implicated by experiments in overexpressing cells treated with both ACV and a BCRP inhibitor, which resulted in the accumulation of the antiviral drug [Article:21859328]. Similarly, multidrug-resistance protein 4 (MRP4) (encoded by ABCC4) conferred cytoprotection against GCV and its metabolites on cells stably transduced with HSV thymidine kinase (TK), and expressing the apical membrane transporter, by accentuating their efflux [Article:12105214]. This observation was confirmed in another study that reported a significantly higher accumulation of GCV (+290%, P < 0.0001) in cells expressing a nonfunctional variant (rs11568658) of ABCC4 and exhibiting a total loss of transport activity when compared to cells expressing the normal protein [Article:27402191].
Toxicity
Nephrotoxicity is the main adverse effect of ACV. It is related to the route of elimination of the drug, being caused by the accumulation of non-soluble drug crystals in the renal tubules, especially in the distal tubular lumen [Article:3376977]. High-dose regimens or misuse by administering ACV too fast as an I.V. bolus are the main cause of renal injury, with ACV-associated crystalline renal failure occurring in 12%–48% of patients treated with the drug [Articles:6285709, 6350472, 3881542]. Renal toxicity is evidenced by an increase in serum creatinine, the appearance of urine sediment, or signs of acute renal failure. Tubular obstruction may be prevented by setting up a high urine flow, reducing the drug-infusion rate, and avoiding associations with other nephrotoxic agents. Repetitive hemodialysis can be used as a treatment [Articles:8416680, 28509218]. Additionally, the accumulation of the aldehyde intermediate of acyclovir in human renal tubular cells in vitro, together with the restoration of cell viability after inhibition of the class I alcohol dehydrogenase suggested that the metabolite was directly involved in kidney damage [Article:22005269].
Neurotoxicity is another important but less frequent side effect of ACV and VACV. It is characterized by neurologic and psychiatric symptoms such as confusion, lethargy, hallucinations, and seizures [Article:2862533]. The mechanisms of ACV neurotoxicity are poorly understood, but it is associated with renal impairment and advanced age (>65 years) [Articles:8038467, 9475829, 34146428], which are responsible for prolonging the half-life of ACV. In addition, the concentrations of CMMG were significantly higher in the plasma of 93 patients who had neurologic side effects than in the plasma of patients without such symptoms (N = 49 vs 44, CMMG concentration = 34.1 ± 39.4 mmol/µL and 4.7 ± 4.7 mmol/µL, respectively, P < 0.001) [Article:12748346]. As expected, renal insufficiency was an aggravating factor for these patients with neuropsychiatric symptoms, 93% of whom had renal impairment. CMMG was subsequently detected in the cerebrospinal fluid (CSF) of nine patients with neurologic symptoms (at a median concentration of 1 µM) but was undetectable in the CSF of 12 patients without symptoms (P < 0.001) [Article:16540518]. This confirmed the role of CMMG in ACV neurotoxicity and reinforced its potential as a toxicity biomarker (with CMMG plasma concentration detection having a sensitivity of 91% and a specificity of 93% [Article:12748346]). In earlier clinical trials, GCV hematologic toxicity was characterized by neutropenia (in 42% of the patients) and thrombocytopenia (in 19%), which were reversible when treatment was discontinued [Article:2847286]. Almost 50% of patients receiving a hematopoietic stem cell transplant (HSCT) and 10% of solid organ transplant (SOT) recipients will exhibit bone marrow myelosuppression [Article:23607363]. No toxic metabolites have been identified for GCV, and its toxicity probably results from its ability to impair DNA replication in hematopoietic progenitors in a dose-dependent manner [Article:3495235]. Creatinine levels may increase during GCV treatment without causing acute impairment of renal function. Neurotoxicity is a less common but significant side effect of GCV treatment. Observed encephalopathy was related to high plasma levels of GCV that were safely and effectively removed by hemodialysis, allowing the patient to recover completely [Article:24403897].
Drug–drug interactions
Drug-drug interactions (DDIs) have been described for ACV and GCV and were followed by nephrotoxicity or myelosuppression for both drugs, and even seizures for GCV. They depend mainly on the competition of co-administrated drugs with ACV or GCV on the transporters involved in the tubular secretion of the antivirals. This may be followed by a decrease in their renal elimination, exposing patients to a risk of toxicity. For example, it has been shown that probenecid (a uric acid reducer) increased the AUC of ACV when these drugs are taken together (AUC0-∞ = 20.5 versus 28.7 µg.h.ml-1), likely by inhibiting the OAT1 transporters [Articles:7103460, 11861798]. Similarly, the co-administration of GCV with probenecid decreased the clearance (-20%) of the antiviral by the renal tubule [Article:9495222].The association of GCV with the combination of antibiotics imipenem and cilastatin is not recommended because of the risk of seizure following the co-administration of these drugs [Article:9475829]. Leukopenia and thrombocytopenia were more frequently observed in transplant patients who received GCV and the immunosuppressant mycophenolate mofetil, and neutropenia was observed in patients with AIDS treated with the antiretroviral drugs zidovudine and didanosine [Articles:8381032, 20720240]. Overall, special care is required when using nephrotoxic drugs that may impair the renal function and therefore, induce higher plasma levels of GCV or ACV.
Therapeutic drug monitoring for acyclovir and ganciclovir
The large panel of possible doses of ACV and GCV according to clinical indication is consistent with the high variability in patients’ exposure. However, therapeutic drug monitoring (TDM) of GCV or ACV has been used to optimize therapy only in specific situations, and the lack of information on dose–concentration relations makes the determination of a clear dose range difficult. TDM for GCV remains controversial despite the need to optimize dosing to avoid toxicity and antiviral resistance [Article:34610621]. In a retrospective cohort of 82 adults with CMV infection who were treated with I.V. GCV (at a median dose of 5 mg/kg for 19 days), the dose was adjusted based on GCV serum concentrations for 17 patients. Overall, no significant associations were observed between viral load or improvement in symptoms and plasma trough concentration (P = 0.20) or peak concentration (P = 0.14) [Article:30602515]. Additionally, no relevant association with GCV hematologic toxicity, neurotoxicity, or nephrotoxicity was observed, leading to the conclusion that TDM for GCV was of limited value for normal adult patients. Conversely, a recent study found that a decrease in the white blood cell count was associated with the highest plasma minimal concentration (Cmin) of GCV (P = 0.007) [Article:34160036]. Greater variability was observed in the subgroup of SOT recipients, as compared with HSCT recipients. Finally, a correlation was found between the intracellular concentration of GCV triphosphate (GCV-TP), the active metabolite of ganciclovir, and the decrease in neutrophils after 3 months of treatment (P < 0.01) [Article:26538506]. There has been more interest in concentration-based dose adaptation for GCV in specific populations such as pediatric patients; however, more studies are needed in the pediatric population. The GCV AUC was found to be more relevant for estimating exposure in children [Article:34749667]. This was demonstrated in the case of a child aged 3.6 years who experienced CMV reactivation after undergoing allogeneic stem cell transplantation while underexposed to GCV and who presented with a high viral load. After the AUC0–12h was monitored and the patient’s creatinine clearance estimated, the dosage was doubled (from 5 mg/kg/12 h to 10 mg/kg/12 h) despite the potential hematologic toxicity, whereupon the viral load started to decrease quickly and soon became undetectable. Similarly, the ACV dose was adjusted in neonates with disseminated HSV-1 infection and acute renal failure by concomitantly monitoring the ACV serum concentrations and viral load to enable the dose to be increased from 1.5 times the standard dose, thereby optimizing the efficacy of the antiviral treatment [Article:25556960].
Pharmacodynamics
Mechanism of action
CMV, HSV-1, HSV-2, and VZV are members of the Herpesviridae family of viruses with double-stranded DNA genomes. CMV targets a wide range of cell types, including endothelial cells, epithelial cells, fibroblasts, smooth muscle cells, leukocytes, and monocytes, and causes multiorgan infections [Article:31336680]. HSV-1 and HSV-2 cause oral and genital herpes, and VZV induces skin lesions (during primoinfection) and zoster (after reactivation). All of these viruses exhibit neurotropism [Article:18351291]. ACV and GCV exert their antiviral activity in host cells after being selectively phosphorylated into monophosphate metabolites (ACV-MP and GCV-MP) by the viral TK of HSV or the ORF47 protein kinase of VZV [Article:6285736]. In CMV-infected cells, a specific phosphotransferase (UL97) takes ACV and GCV in charge [Articles:1319559, 10428917], with GCV being 25 times more potent than ACV against CMV [Article:3032503]. Expression of these viral proteins is the limiting step for the antiviral effect of ACV and GCV and constrains it to infected cells. These monophosphate metabolites are further transformed by the intracellular guanylate kinase (GUK1) into the diphosphate derivatives ACV-DP and GCV-DP [Articles:6248551, 16690197]. The final active triphosphate forms (ACV-TP and GCV-TP) are produced by various cellular kinases that are physiologically involved in phosphorylating guanosine-derived nucleotides to produce GTP and deoxy-GTP; these kinases include phosphoglycerate kinase, pyruvate kinase, phosphoenolpyruvate carboxykinase, nucleoside diphosphate kinase, succinyl-CoA synthetase, creatine kinase, and adenylosuccinate synthetase [Article:7159465]. Their dGTP-like structure makes them the preferential substrates for the viral DNA polymerases (UL54 for CMV and the UL30 catalytic subunit and UL42 for HSV), and the triphosphate forms have a higher affinity for the viral DNA polymerase than for the host-cell DNA polymerase, with this affinity being almost 30 times higher with ACV-TP [Article:6086640]. The incorporation of the drug triphosphates in place of dGTP destabilizes the DNA strands and leads to the suppression of DNA elongation [Articles:7192534, 6285736, 2553730, 8786764].
Clinical indications for GCV and ACV
CMV infection (including retinitis, gastrointestinal infection, pneumonia, and nervous system infection) is an important concern for immunocompromised patients, including those who are HIV positive or transplant recipients [Article:8786764]. As a potential cause of graft failure or complications, the CMV serological status of both the donor and the recipient is systematically determined by measuring CMV-specific immunoglobulin G (IgG) before the transplant is performed and afterwards if the recipient is CMV seronegative. Quantitative DNA amplification by polymerase chain reaction (qPCR) is the gold standard for diagnosing and monitoring CMV infection and disease [Article:29596116]. VGCV is the most current antiviral drug used for CMV prophylaxis because it is suitable for oral use and its efficacy is equivalent to that of GCV. For anti-CMV prophylaxis, VGCV is administered as 900 mg once daily. VACV can be used for prophylaxis in renal transplant patients at a dose of 2000 mg, four times per day, if renal function is normal (with creatinine clearance > 70 mL/min) [Article:29596116]. Because GCV has a more potent inhibitory effect on CMV than does ACV, intravenous GCV is the first-line antiviral recommended for treating proven CMV infections at a dose of 5 mg/kg every 12 hours, with this interval being extended to 24 hours during the maintenance phase. In CMV-positive HSCT recipients (representing 30%–80% of HSCT patients, depending on donor and recipient status), intravenous GCV (5 mg/kg/12 h) will be used to treat post-transplant infection or reactivation [Article:33515202]. Finally, GCV (6 mg/kg/12 h) and VGCV (currently under study [Article:33964331]) are also indicated for treating congenital CMV infection, which affects 0.5%–0.7% of neonates in the United States each year, with most being asymptomatic [Article:24257422]. Compared to GCV, ACV has a wider activity spectrum against herpesviruses (HSV-1, HSV-2, and VZV), and it is available in various pharmaceutical forms (oral tablets, capsules, suspensions, topical creams, or a solution for infusion). The ACV dose range for HSV-1 and HSV-2 genital infections is 800–1600 mg/day until complete recovery, and the specific dose depends on the stage of infection (primoinfection, recurrent disease, etc.) . VACV, with its improved bioavailability, enables the frequency of drug intake to be reduced. In cases of severe disease and systemic complications, ACV is administered by infusion at a dose of 5–10 mg/kg every 8 hours until there is improvement , and it is administered at up to 60 mg/kg every 8 hours for 14–21 days for herpes congenital infection [Article:34030813]. Finally, oral ACV at 20 mg/kg four times a day for 5 days was shown to reduce the complications of chickenpox when administered 24 hours after the rash appeared [Article:1944438].
Viral resistance
Resistance to ACV or GCV therapy may result from quantitative or qualitative changes in viral enzymes and/or the targeted proteins. Mutations in the HSV TK gene UL23 were found in patient isolates exhibiting clinical ACV resistance, and some canonical mutations (M460V/I, H520Q, C592G, A594V, L595S, and C603W) in the UL97 gene of CMV have been related to antiviral resistance [Articles:22001948, 21078929, 23198963]. Importantly, an isolate from a recipient can harbor multiple mutations or various virus subpopulations with different mutations. Additionally, mutations in the UL54 gene encoding the targeted viral DNA polymerase have been shown to be a consequence of prolonged exposure (for weeks to months) to antiviral therapy [Article:22001948]. Finally, triphosphate metabolites are inactivated by the NUDT15 (nucleoside diphosphate–linked moiety X-type motif 15) diphosphatase, which converts them into their respective monophosphate derivatives and limits their antiviral effect against CMV in vitro [Articles:34234136, 34192523].
Pharmacogenomics
The association of NUDT15 genetic variant (rs116855232, NM_018283.2:c.415C>T, R139C, or the *3 allele), was first identified in a genome-wide association study (GWAS) of Korean patients treated with azathioprine for Crohn disease in 2014 [Article:25108385]. A year later, another GWAS found that the same variant was related to thiopurine intolerance in pediatric patients with acute lymphoblastic leukemia [Article:25624441]. Since then, additional variants have been described, and some NUDT15 polymorphisms—including rs746071566–rs116855232 (the *2 allele), rs147390019 (the *4 allele), and rs186364861 (the *5 allele)—may alter thiopurine metabolism, especially in the Asian and Hispanic populations. Indeed, a cumulative frequency of 10.9% was found in a cohort of 55 pediatric patients harboring the *2, *3, or *5 alleles [Article:28445187]. In these patients, the accumulation of thioguanine nucleotides (TGNs), which are incorporated into DNA (as DNA-TG) to induce cell apoptosis, correlated inversely with the number of NUDT15 risk alleles (P < 0.001), highlighting the increased conversion of TGN to DNA-TG, which increases the risk of toxicity, as previously observed in NUDT15 knockout cells [Article:26878724]. Consequently, the Clinical Pharmacogenetics Implementation Consortium (CPIC) defined four NUDT15 activity groups based on the patient’s diplotype determination: normal (*1/*1), intermediate (*1/*2, *1/*3, *1/*4, and *1/*5) and possible intermediate (*2/*5 and *3/*6), and poor (*2/*2, *2/*3, *3/*3, and *5/*5) metabolizers [Article:30447069]. Overall, this critical role of NUDT15 in the thiopurine metabolism pathway suggested that the enzyme could metabolize additional purine-derivative drugs. An in vitro drug-screening study identified ACV-TP and GCV-TP as substrates and showed that NUDT15 deficiency potentiates the antiviral effect against CMV and cytotoxicity against host hematopoietic cells [Article:34234136]. In the same study, the degree of loss of NUDT15 activity in both host tissue and donor cells was associated with a correspondingly improved anti-CMV clinical response to ACV prophylaxis in 248 HSCT recipients (P = 0.015). Low NUDT15 activity in CMV-positive donor cells was associated with an increased rate of graft failure (P = 0.047), suggesting a protective role for NUDT15 against ACV and/or GCV cytotoxicity. Recently, an NUDT15-linked severe neutropenia was reported in an Indian patient who received azathioprine (100 mg/day) and VGCV (450 mg/day) as CMV prophylaxis after a kidney transplant [Article:35064735]. Genotyping revealed the patient to be a normal metabolizer for TPMT (thiomethylpurine transferase), the main enzyme that catabolizes azathioprine, but the carrier of two copies of the *3 allele of NUDT15. Azathioprine treatment was associated with early severe myelosuppression. This subsequently abated, but a second isolated episode of neutropenia after VGCV was reintroduced helped attribute this toxicity to the antiviral. The ABCC4 NM_001105515.2:c.559G>T (rs11568658, G187W) polymorphism was associated with a decrease in neutrophil count in a group of 174 renal transplant patients receiving VGCV (P = 0.0078) and led to the accumulation of GCV in cells by abolishing its efflux [Article:27402191]. Similarly, a retrospective study that included 134 heart transplant recipients showed that a variant in the gene encoding hepatocyte nuclear factor 1 homeobox A, HNF1A 5102A>C (rs1169288, I27L), was associated with a higher rate of drug-induced leukopenia in the first 6 months post-transplant in patients receiving GCV or VGCV and/or mycophenolate immunosuppressant (odds ratio = 6.19; 95% confidence interval (CI) = 1.97–19.43, adjusted P = 0.002) [Article:34253456]. Twelve months after transplant, 46 patients in the cohort developed drug-induced leukopenia, and SNPs in HNF1A (rs1169288 5102A>C, rs2393791 T>C), SLC13A1 (solute carrier family 13 member 1, rs2140516 T>C), and MBOAT1 (membrane bound O-acyltransferase domain containing 1) were suggested to be associated with this outcome (P < 0.05). As ALDH2 is essential for transforming ACV to its CMMG toxic metabolite, the influence of ALDH2 genetics was assessed in 18 Japanese patients with end-stage renal disease who were treated with oral VACV [Article:18974607]. However, the only significant relation observed was one between reduced function of the ALDH2 enzyme (caused by the *2 allele, rs671, G504L) and prolongation of ACV half-life; there were no other associations with the pharmacokinetic parameters of ACV or with those of its CMMG metabolite. The analysis of 117 candidate variants in a cohort of 2169 CMV-seropositive HSCT recipients showed that ABCB1 rs1045642 (3435A>G, a minor allele associated with decreased efflux) in donors was associated with a higher risk of any CMV reactivation in recipients (hazard ratio = 0.79, 95% CI = 0.6–1, for AG–GG versus AA, P < 0.03 in the replication cohort) [Article:34269803]. The researchers hypothesized that decreased efflux of immunosuppressants (cyclosporine or tacrolimus) leads to the inhibition of lymphocyte function and to an increased probability of CMV reactivation.
Conclusion
ACV and GCV have been used for many years for the treatment and prophylaxis of herpesvirus infections, including HSV-1, HSV-2, VZV, and CMV infections. Significant intraindividual and interindividual variability was soon observed in patients, leading to considerable discrepancies in terms of efficacy and toxicity: excessive drug exposure may induce significant side effects, whereas suboptimal exposure can lead to the resurgence of viral resistance. The lack of robustness of many studies has prevented the implementation of an optimal therapeutic window, and only those studies constrained to a specific population have demonstrated the potential benefit of using TDM. Apart from the PK–PD relations, attention should be given to ACV and GCV pharmacogenomics, of which only a few studies have been published. The findings regarding NUDT15 and ABCC4 variants are consistent with cellular proteins playing a metabolic role in GCV and ACV disposition, making those drugs good candidates for pharmacogenomics-driven therapy. Further studies combining patients' clinical data, pharmacogenomics investigations, and functional assays are needed to decipher this variability with respect to ACV and GCV, with the aim of optimizing antiviral therapy.
Reactions & interactions (37)
-
Biochemical Reaction
acyclovir triphosphate → acyclovir monophosphate
-
Biochemical Reaction
acyclovir diphosphate → acyclovir triphosphate
-
Biochemical Reaction
aciclovir → acyclovir aldehyde
-
Biochemical Reaction
aciclovir → acyclovir monophosphate
-
Biochemical Reaction
ganciclovir → Ganciclovir Monophosphate
-
Biochemical Reaction
acyclovir monophosphate → acyclovir diphosphate
-
Biochemical Reaction
Ganciclovir Monophosphate → ganciclovir diphosphate
-
Biochemical Reaction
acyclovir aldehyde → carboxymethoxymethylguanine
-
Biochemical Reaction
aciclovir → 8-hydroxyacyclovir
-
Biochemical Reaction
ganciclovir triphosphate → Ganciclovir Monophosphate
-
Biochemical Reaction
ganciclovir diphosphate → ganciclovir triphosphate
-
Catalysis
NUDT15 → Biochemical Reaction
-
Catalysis
SLC47A2 → Transport
-
Catalysis
SLC47A1 → Transport
-
Catalysis
ABCC4 → Transport
-
Catalysis
SLC47A1 → Transport
-
Catalysis
SLC47A2 → Transport
-
Catalysis
ABCG2 → Transport
-
Catalysis
ADH1A → Biochemical Reaction
-
Catalysis
GUK1 → Biochemical Reaction
-
Catalysis
SLC22A8 → Transport
-
Catalysis
SLC22A1 → Transport
-
Catalysis
SLC22A6 → Transport
-
Catalysis
SLC22A7 → Transport
-
Catalysis
SLC22A6 → Transport
-
Catalysis
SLC22A1 → Transport
-
Catalysis
SLC22A7 → Transport
-
Catalysis
GUK1 → Biochemical Reaction
-
Catalysis
ALDH2 → Biochemical Reaction
-
Catalysis
AOX1 → Biochemical Reaction
-
Catalysis
NUDT15 → Biochemical Reaction
-
Inhibition
acyclovir triphosphate → Viral DNA Polymerase
-
Inhibition
ganciclovir triphosphate → Viral DNA Polymerase
-
Transport
ganciclovir → ganciclovir
-
Transport
aciclovir → aciclovir
-
Transport
aciclovir → aciclovir
-
Transport
ganciclovir → ganciclovir
Edit history (1)
- 2022-03-10 Create