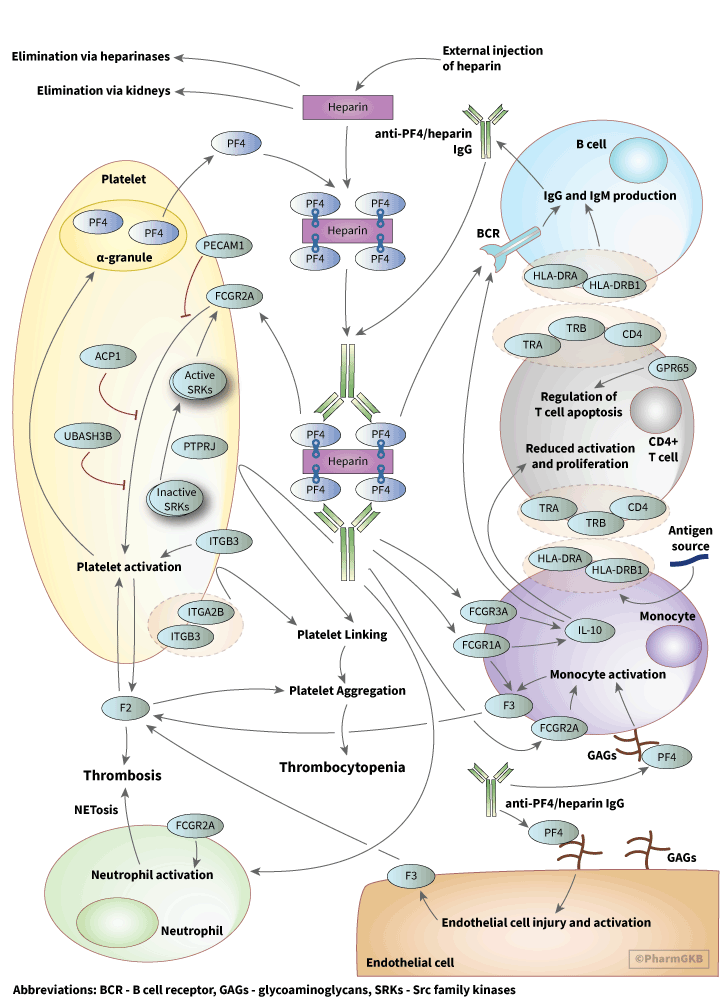
About this pathway
Introduction
Unfractionated heparin (UFH) and low molecular weight heparin (LMWH) are mainstays of therapeutic anticoagulation with multiple desirable pharmacologic traits and a wealth of evidence supporting their use [Article:22315270]. UFH and LMWH have the potential to cause heparin-induced thrombocytopenia (HIT), an off-target, immune-mediated adverse drug reaction (ADR). HIT occurs in up to 2.4% of patients treated with heparin anticoagulants, has a mortality rate as high as 30%, and may result in catastrophic thromboembolic complications, including life- and limb-threatening thrombosis [Articles:12480713, 16202170, 16113796, 15985543]. Considering the widespread use of these agents and the severity of this reaction, the inability to predict HIT constitutes a considerable liability for heparin anticoagulant treatment. Genetic variation has the potential to constitute clinically implementable biomarkers that predict ADRs such as HIT, improving the safety of heparin anticoagulants [Articles:22315264, 30398086]. Previous studies aimed at identifying genetic influences on HIT are primarily candidate gene studies that have focused on polymorphisms in FCGR2A and the HLA locus. Genome-wide association studies (GWAS) have also been conducted with conflicting results. Here we review the pathophysiology of HIT, the likely biological pathways influencing HIT risk, and the evidence of genetic associations with HIT. As both UFH and LMWH have the potential to cause HIT, this summary will focus on UFH due to its higher HIT risk [Article:22315270].
Heparin Induced Thrombocytopenia
Treatment with heparin anticoagulants can result in two types of drug-induced thrombocytopenia. Type I HIT is a non-immune mediated reaction where few clinical implications are present and platelets stabilize quickly. Type I HIT is more common and can occur as early as one day into therapy. Type II HIT is immune-mediated, less common, and typically occurs 5-14 days after initial therapy. For the purposes of this description, HIT is used to refer to Type II HIT.
The pathogenesis of HIT involves binding of the linear polyanion heparin to the positively charged protein platelet factor 4 (PF4), an endogenous chemokine stored in alpha granules of platelets, as shown in the pathway image above. PF4 is involved in bacterial defense through interactions with bacterial lipopolysaccharides suggesting that HIT may be a misdirected response towards exogenous heparins [Articles:21659541, 22942185, 26176382]. Complexes of PF4 and heparin are recognized by IgG antibodies, forming PF4/heparin-IgG complexes [Article:1718005]. Interactions between PF4 and heparin are dependent on charge and stoichiometric ratios; excess of either PF4 or heparin will prevent assembly of antibody complexes [Articles:15304392, 17848616]. These antibody complexes activate platelets and monocytes via the platelet Fcγ-receptor (FcγRIIa), encoded by the gene FCGR2A, resulting in thrombin generation and thrombotic complications [Articles:3416077, 24671506, 26176382]. Recent data suggest that release of Neutrophil Extracellular Traps (NETosis) following neutrophil activation via the FcγRIIa receptor is a causative factor in heparin induced thrombocytopenia-associated thrombosis (HITT) [Article:30899022]. In macrophages, FCGR1A and FCGR3A have been linked to release of IL-10, an important immunoregulatory cytokine that may have relevant downstream effects in HIT [Articles:12782719, 32046249]. Current models for HIT indicate that a yet unidentified antigen exposure happens prior to heparin exposure, creating a primary immune response that primes production of relevant IgGs.
Platelet monitoring is a primary method of detecting HIT during heparin treatment. American College of Chest Physician (CHEST) guidelines recommend for patients with a greater than 1% chance of developing HIT to have their platelet count checked every 2-3 days in the first 14 days of treatment with heparin [Article:22315270]. Scoring systems such as the 4Ts score, which uses clinical characteristics to estimate the likelihood of HIT prior to laboratory confirmation, are useful in risk stratification of patients based on HIT risk [Articles:20149589, 22990018]. American Society of Hematology recommendations include assay testing for HIT if patients have an intermediate to high 4Ts score [Article:30482768]. Enzyme-linked immunosorbent assays are used to detect PF4/heparin antibodies and screen for HIT in suspected patients. Up to 50% of heparin-treated patients will develop PF4/heparin antibodies, but only a fraction of those patients will develop HIT and its thromboembolic complications [Articles:7715641, 9674736, 10330384]. Therefore, platelet reactivity testing using highly sensitive and specific functional assays is necessary to diagnose HIT. Such assays include the Serotonin Release Assay (SRA) and the Heparin Induced Platelet Activation Assays (HIPA) [Article:28398258]. These tests are technically demanding, usually restricted to specialized laboratories, and have the potential to result in significant delays in diagnosis. Clinical approaches to functional assay-confirmed HIT focus on management and treatment of the condition after thrombocytopenia or other signs of HIT are presented. Discontinuation of heparin anticoagulants and initiating a non-heparin anticoagulant is critical to prevent HIT-related thrombosis. HIT patients that manifest thrombotic complications should be transitioned to non-heparinoid anticoagulant such as argatroban, lepirudin, danaparoid, or bivalirudin.
Patients may present with a HIT-like syndrome termed autoimmune HIT without heparin exposure [Article:28846826]. Like HIT, autoimmune HIT results from platelet activation by PF4/heparin-like antibodies, but in autoimmune HIT these antibodies form in response to PF4 binding polyanionic compounds other than heparin. Other polyanions such as RNA, proteoglycans, polyphosphates, and bacterial cell wall components may bind to PF4 to stimulate an immune response, providing multiple pathways for autoimmune HIT. Treatment often includes aggressive therapy with an anticoagulant other than heparin (as heparin can exacerbate the condition) until platelet counts return to normal. Similar clinical features to autoimmune HIT have been observed in cases of vaccine-induced thrombotic thrombocytopenia (VITT) due to adenoviral vector SARS-CoV-2 vaccines[Articles:33861524, 33835769, 34051613, 33861525, 34108714, 34010527]. VITT is characterized by high levels of antibodies to PF4-polyanion complexes, unusual thrombotic sites, and high mortality, bearing striking similarity to autoimmune HIT. Clear similarities between Coronavirus Disease 2019 (COVID-19) and HIT have been described in detail, including a high rate of PF4/heparin antibodies and increased risk of HIT in COVID-19 patients [Articles:32990022, 32697931, 32810772, 32658337, 32905282, 32841919, 33639037]. Rates of thrombocytopenia in COVID-19 patients are estimated to be 57.7% in severe cases of infection and 31.6% in non-severe cases, although different studies have found varying rates [Article:32984764].
Heparin Pharmacology
Heparins are produced by all mammals in basophils and mast cells, but exogenous heparins are used for therapeutic and prophylactic anticoagulation. Heparin is often supplied in an unfractionated form, UFH, which is used in the inpatient setting due to ease of administration, low cost, immediate onset of action, short half life, availability of lab monitoring, reversibility, a lack of renal adjustments, and a wealth of evidence supporting its use [Articles:22315264, 30398086]. Clinical monitoring of UFH can be done via anti-factor Xa testing or by proxy using activated partial thromboplastin time (aPTT) testing [Article:26780745]. UFH contains sulfated glycosaminoglycans of variable length that bind to antithrombin III, a serine protease. Heparin binding to antithrombin III induces an activating conformational change that allows it to inactivate other clotting factors, including Factors IIa (thrombin) and Xa. Heparins have a minimum five saccharide chain length and simultaneously bind to both antithrombin III and thrombin if the chain length is 18 or greater [Article:6721831], as shown below. LMWHs, such as dalteparin and enoxaparin, have been fractionated to fewer chain lengths and have a lower potential to cause HIT. A synthetic heparin pentasaccharide, fondaparinux, has a still lower potential for HIT.
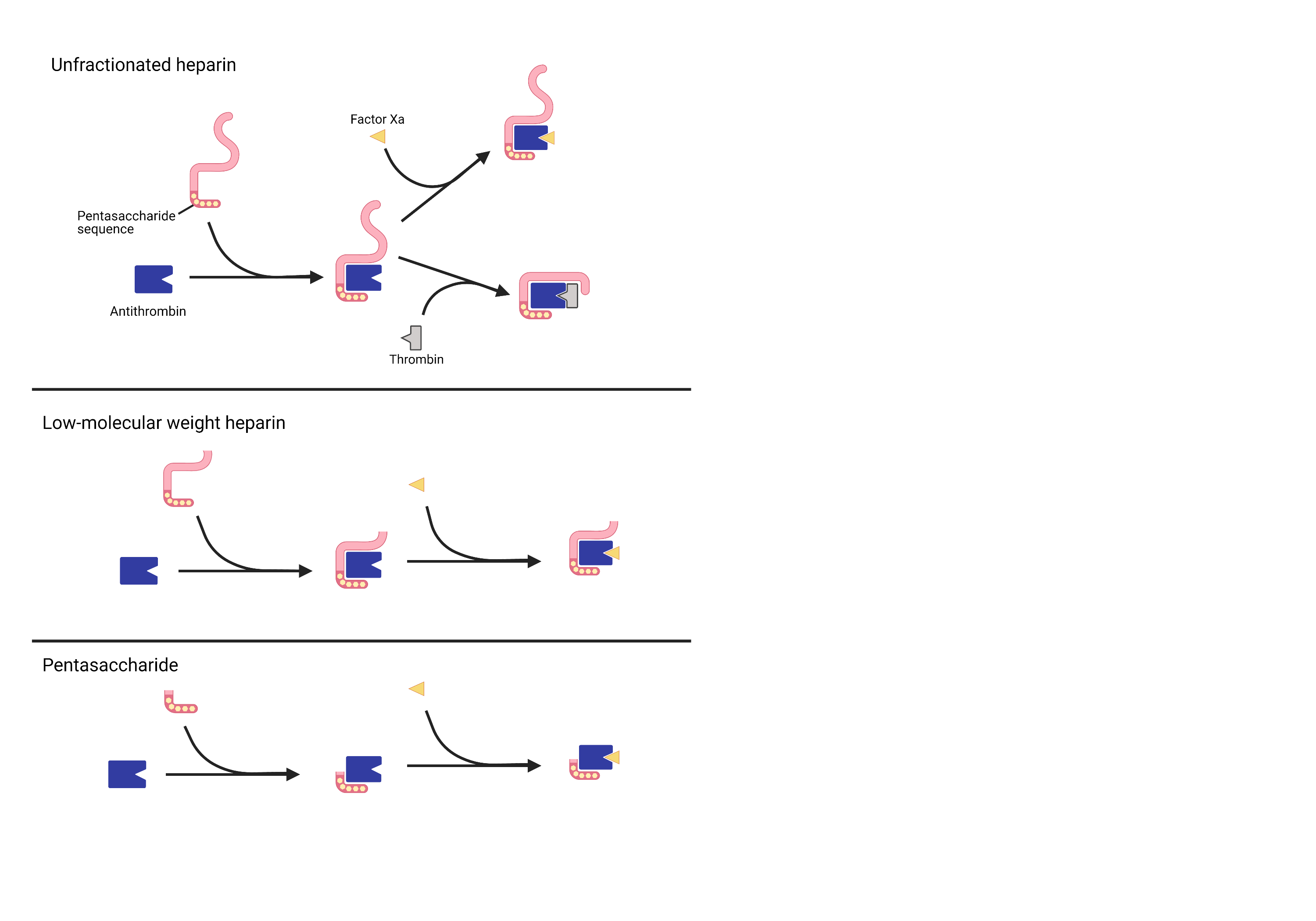 The variable chain lengths of (a) unfractionated heparin, (b) enoxaparin, and (c) a pentamer such as fondaparinux offer different pharmacologic profiles, including differences in binding to antithrombin and subsequent inactivation of Thrombin and Factor Xa.
The variable chain lengths of (a) unfractionated heparin, (b) enoxaparin, and (c) a pentamer such as fondaparinux offer different pharmacologic profiles, including differences in binding to antithrombin and subsequent inactivation of Thrombin and Factor Xa.
Heparin Pharmacokinetics
UFH and exogenous heparins do not behave as their endogenous counterparts. The pharmacologic profile of UFH is complex due to its nature as a linear glycosaminoglycan of variable length [Article:26672027]. UFH is typically isolated from bovine lung or porcine intestine and this results in variability in chain lengths, product strengths, and patient response, requiring the use of therapeutic monitoring for UFH. Increasing UFH dose is also a risk factor for HIT [Article:22315270]. UFH can be modeled using non-linear Michaelis-Menten kinetics. UFH can quickly bind to circulating PF4 in the blood as well as other proteins, including albumin, plasmin, and fibrinogen. UFH distributes throughout the blood compartment in the intravascular space and is protein bound to a clinically relevant extent [Article:1505142]. For the purposes of modeling, UFH can be considered to have a volume of distribution limited to the intravascular space. Two-compartment models have also been determined [Article:25638272].
UFH has a half-life of effect on clotting time of approximately 1.5 hours [Articles:13819631, 808390]. However, half-life of the molecule is variable depending on the dose given [Articles:808390, 512841]. There appear to be two processes by which heparin is eliminated. A saturable mechanism which predominates at lower doses, and a non-saturable mechanism at higher doses [Article:1505142]. In the saturable mechanism, heparins are phagocytized by the reticuloendothelial system, which plays a large role in elimination [Articles:13197556, 512841]. The non-saturable mechanism of elimination is via renal excretion. Heparins are also capable of being depolymerized via Heparanase-1 and via N-desulfation [Articles:13819631, 7107605, 3159732, 18023704].
Pharmacogenetics of Heparin Induced Thrombocytopenia
Pharmacogenomics is an area of great interest in HIT research as predictive genetic markers might have clinical utility. A number of pharmacogenetic associations have been found regarding HIT, but few of these studies have been rigorously replicated [Articles:8555090, 9716579, 11255285, 15191947, 18208809, 22239992, 22677127, 22793995, 23621699, 25503805, 25680756, 28688202, 29934777]. The majority of the associated genes are related to the immune system.
Fcγ receptors, especially FcγRIIA and FcγRIIIA (encoded by FCGR2A and FCGR3A, respectively), are frequently discussed in HIT pharmacogenomic research. These receptors are a family of immunoglobulin proteins that bind to antigen-antibody complexes including PF4/heparin–IgG complexes involved in HIT. The H131R polymorphism of the FCGR2A (CD32A) gene (rs1801274) was first associated with increased HITT in a study with 20 HIT patients and 24 healthy controls [Article:8555090]. This association was further supported in a larger study with HIPA-confirmed HIT patients confirmed and non-HIT thrombocytopenia or thrombosis patients that tested negative for PF4/heparin antibodies (Abneg) [Article:8555090]. Further research has found that individuals homozygous for the H131R polymorphism were at significantly higher risk of thrombosis, likely due to increased platelet activation by antibodies [Article:25680756]. The consistent results of these studies have provided strong evidence for a role of FCGR2A-H131R in HITT risk.
Research has also expanded into factors that may influence FCGR2A expression, such as low-molecular-weight phosphotyrosine phosphatase (LMW-PTP) and variation in protein tyrosine phosphatase (PTPRJ). ACP1 encodes LMW-PTP, which is involved in phosphorylation of FCGR2A in platelets [Article:17537991]. In a non-replicated study with confirmed HIT patients, high catalytic efficiency ACP1 haplotypes were more common in HIT patients [Article:23621699]. The authors reasoned that this may be attributable to the fact LMW-PTP regulates ZAP-70, which plays a critical role in T-cell development and lymphocyte activation. However, there was no difference in genotypic frequency between HIT and Abpos patients, suggesting that ACP1 polymorphisms do not play a role in HIT antibody pathogenicity [Article:23621699]. In one study, patients homozygous for Q276P (rs1566734) and R326Q (rs1503185) in PTPRJ were found to be underrepresented in HIT cases, suggesting that homozygotes are more resistant to HIT development [Article:22677127]. In addition, individuals homozygous for FCGR3A-F158V (rs396991) were more likely to be diagnosed with HIT [Article:15191947].
Several other genetic associations with HIT have been identified, although additional research is warranted to confirm a true effect. A polymorphism in ITGB3 (glycoprotein IIIa subunit, GPIIb/IIIa is a fibrinogen receptor on platelets involved in activation) called human platelet antigen 1b (HPA-1b, L33P, rs5918) and PECAM1-L125V (platelet endothelial cell adhesion molecule) have been associated with increased thrombosis in HIT patients [Article:22793995]. The G20 allele of the IL10G microsatellite was found to influence immune response to heparin and antibody production and an intronic SNP on chromosome 5 with no known function (rs1433265) was strongly associated with HIT in a GWAS [Article:29934777]. One study determined that Human Leukocyte Antigens (HLA) play a role in HIT pathogenesis, especially HLA-DR, although the study had very small sample size [Article:11255285]. A later study examined HLA gene variants, as these can be biomarkers of immune-mediated adverse drug reactions and determined that HLA-DRB3*01:01 is a potential risk factor for HIT, although the population examined was mostly of European descent [Article:28688202]. No replication was performed for any of these studies, so additional research is needed to confirm these associations. A GWAS with a discovery cohort and replication cohort observed a statistically significant association between GPR65, also known as T-Cell Death-Associated Gene 8 (TDAG8), and HIT [Article:25503805]. In the discovery cohort, GPR65 SNPs were significantly associated with HIT and in the replication cohort, GPR65 SNPs were associated with PF4/heparin antibodies, but not HIT [Article:25503805]. This may be caused by the small sample size or differences in HIT case diagnosis, as the replication cohort had more stringent criteria for inclusion in the HIT group. Additionally, HIT patients in the discovery cohort were not functionally confirmed and the study was unable to differentiate predictors of antibody response and HIT. GPR65 is connected to regulation of T cell apoptosis and immune-mediated inflammation via reduction of cytokine production in T cells and macrophages, which are both important players in HIT [Articles:8599842, 22445881]. GPR65 was also shown to reduce the severity of two hypersensitivity models in mice, but the precise role of GPR65 in HIT remains unknown [Article:21238451].
Conclusion
HIT is a rare but potentially fatal condition induced by heparin anticoagulants that are widely used in clinical practice. Genetic polymorphisms have the potential to predict HIT and HITT, and pharmacogenomic research has associated multiple genes with these conditions. FCGRA2-H131R is the most supported by prior studies and has the greatest potential thus far to predict HITT. However, past research has several limitations that may reduce the potential impact of this literature. These include limited sample size, inconsistent phenotype definitions, and differing distinctions between antibody positive patients. While the potential for pre-emptive genetic testing to prevent HIT and HITT exists, more research is necessary to discern the validity and utility of genetic variation in the prevention of HIT and HITT.
Reactions & interactions (47)
-
Activation
FCGR1A → IL10
-
Activation
FCGR3A → IL10
-
Activation
F3 → F2
-
Activation
F3 → F2
-
Activation
anti-heparin/PF4 IgG → PF4
-
Activation
anti-heparin/PF4 IgG → PF4
-
Activation
Heparin/PF4/IgG complex → FCGR2A
-
Activation
FCGR1A → F3
-
Activation
Heparin/PF4/IgG complex → FCGR2A
-
Activation
endothelial cell activation → F3
-
Activation
Src family kinases → FCGR2A
-
Activation
Heparin/PF4/IgG complex → B-cell receptor
-
Activation
monocyte activation → F3
-
Activation
Heparin/PF4/IgG complex → FCGR1A
-
Activation
Heparin/PF4/IgG complex → FCGR3A
-
Activation
Heparin/PF4/IgG complex → FCGR2A
-
Activation
TRA/TRB/CD4 complex → HLA-DRA/HLA-DRB1 complex
-
Activation
IL10 → B-cell receptor
-
Catalysis
PTPRJ → Conversion
-
Comes From
platelet activation → F2
-
Complex Assembly
Heparin/PF4 complex + anti-heparin/PF4 IgG → Heparin/PF4/IgG complex
-
Complex Assembly
PF4 + heparin → Heparin/PF4 complex
-
Conversion
IgG → anti-heparin/PF4 IgG
-
Conversion
Src family kinases → Src family kinases
-
Inhibition
ACP1 → FCGR2A
-
Inhibition
UBASH3B → FCGR2A
-
Inhibition
PECAM1 → FCGR2A
-
Leads To
FCGR2A → platelet activation
-
Leads To
FCGR2A → monocyte activation
-
Leads To
F2 → heparin-induced thrombocytopenia
-
Leads To
GPIIb/IIIa + ITGB3 → platelet aggregation
-
Leads To
GPR65 → regulation of T cell apoptotic process
-
Leads To
F2 → platelet activation
-
Leads To
immunoglobulin production → IgG
-
Leads To
Heparin/PF4/IgG complex → platelet aggregation
-
Leads To
IL10 → negative regulation of activated T cell proliferation
-
Leads To
B-cell receptor → immunoglobulin production
-
Leads To
ITGB3 → platelet activation
-
Leads To
neutrophil activation → Thrombosis
-
Leads To
platelet activation → PF4
-
Leads To
F2 → Thrombosis
-
Leads To
HLA-DRA/HLA-DRB1 complex → immunoglobulin production
-
Leads To
FCGR2A → neutrophil activation
-
Leads To
platelet aggregation → heparin-induced thrombocytopenia
-
Leads To
PF4 → monocyte activation
-
Leads To
PF4 → endothelial cell activation
-
Transport
PF4 → PF4
Edit history (5)
- 2021-09-21 Create
- 2021-10-05 Update Minor edits to text description
- 2022-03-30 Update Added citation PMID
- 2023-02-01 Update changed link for HLA-DRB3
- 2025-09-15 Update Removed broken link