About this pathway
Artemisinin is a phytoconstituent obtained from the Chinese medicinal herb Artemisia annua [Article:19851082]. It has a sesquiterpene lactone structure with an internal peroxide bridge, which provides a different structural prototype compared to classical antimalaria drugs such as e.g. chloroquine, quinine, proguanil or sulfadoxine [Article:15318224], WHO/HTM/MAL/2006.1108.
Artemisinin has a poor bioavailability limiting its effectiveness. Therefore semisynthetic derivatives of artemisinin; artesunate, artemether, and arteether; have been developed [Article:19851082]. Artemisinins are short-acting antimalarial agents used to treat uncomplicated Plasmodium falciparum malaria. They kill parasites more rapidly than conventional antimalarial drugs, and are active against both the sexual and asexual stages of the parasite cycle [Article:18193915]. Due to their short half-life and to prevent development of resistance, artemisinin compounds are often combined with one or two long-acting antimalarial drugs -- amodiaquine, mefloquine, sulfadoxine/pyrimethamine or lumefantrine [Article:19926036] as artemisinin-based combination therapy (ACT). ACT is now being widely used as the first-line treatment for Plasmodium falciparum malaria throughout the world [Article:19641202].
Depending on their chemical structure different formulations are available for artemisinin and its derivatives. For artemisinin a wide variety of formulations for oral, parenteral and rectal use are available. Artemether can be given as an oil-based intramuscular injection or orally. Artesunate can be given orally, rectally or by the intramuscular or intravenous routes. Arteether is oil-based so water insoluble and therefore given by intramuscular injection only [WHO/HTM/MAL/2006.1108]. Factors affecting the metabolism of artemisinin drugs are food intake, gender, and disease state [Article:11069212].
The mechanism of action of artemisinin and its derivatives are not completely defined, however the sarcoplasmic-endoplasmic reticulum ATPase (PfATP6 or PfSERCA) gene of Plasmodium has been suggested as possible parasite-specific target [Articles:12931192, 15533761, 18193915]. This enzyme is thought to be critical for parasite survival, and is damaged by carbon-centered free radicals released during metabolism of the artemisinin peroxide [Article:15318224].
The antimalarial activity of the artemisinin class, as well as their toxicity, is a result of their peroxide bridge. The metabolites therefore can be divided in biologically active hydroxylated compounds with an intact endoperoxide bridge and biologically inactive deoxy metabolites where the peroxide bridge has been reduced to an epoxide [Article:11069212]. Further all these metabolites undergo glucuronidation and are excreted in the urine or feces [Article:11069212]. Dihydroartemisinin appears to be the principal metabolite of all artemisinin derivatives but not artemisinin itself [Article:11069212]. The conversion occurs to a varying degree and through different mechanisms [Article:11069212]. Dihydroartemisinin is an active metabolite and also available as a drug in a fixed drug combination with piperaquine. The elimination half-lives of artemisinins are ranging from 2 to 5 hours for artemisinin, <1 hour for artesunate and 2 to 4 hours for artemether [Article:18193915]. Artemisinins have a favorable safety profile, probably due to their fast elimination [Articles:15533761, 19926036]. However, neurotoxicity has been reported as an adverse effect [Articles:16828992, 19851082]. The extent of the neurotoxicity is dependent on the nature of the compound and its formulation as well on the route of administration [Article:19851082]. Also type 1 hypersensitivity reactions have been reported as adverse reaction to oral artesunate [Article:11355556].
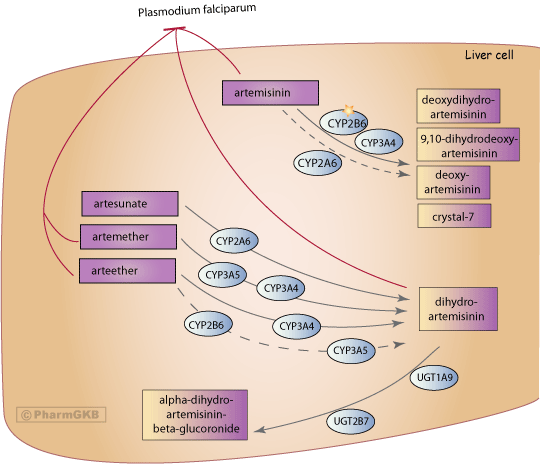
After oral administration of artemisinin, deoxyartemisinin, deoxydihydroartemisinin, 9,10-dihydrodeoxyartemisinin and a metabolite named 'crystal 7' were identified in human urine. All four metabolites are inactive due to the lack of the endoperoxide bridge [Articles:2084705, 8053019, 11069212]. The candidate genes involved in the metabolism of the different artemisinins differ slightly as seen in the figure. Based on in vitro studies in human liver microsomes, the metabolism of artemisinin is primarily mediated by CYP2B6 with a secondary contribution of CYP3A4. It has been suggested that CYP3A4 is probably of importance in individuals with low CYP2B6 expression [Article:10583023]. Artemisinin metabolism can be performed by CYP2A6 in vitro but this is considered to be less relevant in vivo than CYP2B6 and CYP3A4 [Article:10583023]. Administration of the CYP2B6 inhibitor orphenadrine and the CYP3A4 inhibitor ketoconazole to human liver microsomes decreased artemisinin disappearance rates by 75% and 10%, respectively [Article:9728896].
Artesunate is hydrolyzed within minutes to its active metabolite, dihydroartemisinin, which is considered to be responsible for the antimalarial activity [Articles:2084705, 11791963]. In vitro data provide evidence for CYP2A6 as the major metabolizing enzyme for artesunate [Article:12920490]. Artemether is rapidly demethylated to the active metabolite dihydroartemisinin by CYP3A4 and CYP3A5 [Articles:18193915, 11069212]. In vitro studies using human recombinant cytochrome P450 enzymes showed that primarily CYP3A4 is involved in the metabolism of arteether to its active metabolite, dihydroartemisinin. Secondary contributions by CYP2B6 and CYP3A5 were found [Article:9531517]. Metabolism studies using rat liver microsome preparations also identified further minor, inactive metabolites of artemether and arteether [Article:2084705]. Dihydroartemisinin is converted to inactive metabolites via glucuronidation catalyzed by UDP-glucuronosyltransferases, in particular UGT1A9 and UGT2B7 [Article:12167566]. UGT1A1 and UGT1A8 have also been reported to be involved [Article:10496300]. Dihydroartemisinin is also eliminated in bile as minor glucuronides, such as tetrahydrofurano acetate [Article:11791963].
To date the number of studies investigating metabolites of artemisinin and its derivatives in humans are still small [Article:19851082]. In addition these studies are not directly comparable with different drugs, formulations, routes of administration and populations being tested leading to a fragmented picture of metabolism. Depending on the route of administration and the derivative probably further metabolites exist that have yet to be identified. For example, an additional unidentified metabolite with an intact peroxide bridge was detected in the urine of two Chinese volunteers after intravenous administration of artesunate [Article:11069212].
There are currently no comprehensive clinical studies of pharmacogenomics of artemisinins reported in PubMed (02/2010). However, possible interactions of importance can be predicted from understanding the variability of the metabolizing enzymes involved, the frequency of these variations in different populations and the metabolism of co-administered drugs. Although the artemisinin metabolizing enzyme CYP2B6 is one of the most polymorphic CYP genes in humans [Article:17638512] CYP2B6 VIP, no effect on clinical outcome as measured by fever and parasite clearance rates have been seen, possibly due to the use of combination therapy with other antimalarial drugs [Article:19926036]. Well documented variants for CYP3A4, CYP3A5 and CYP2A6 also exist but have not been tested in respect to artemisinins.
Artemisinin, artemether, and dihydroartemisinin have been shown to induce CYP3A activity, in addition artemisinin also up-regulates CYP2C19 and CYP2B6 [Articles:9728896, 10626755, 12844133, 17521300]. CYP2C19 is also activated by arteether [Article:17521300]. This auto-induction was proposed to be the main mechanism causing the decline of plasma concentrations of artemisinins after repeated dosing [Articles:9585803, 12844133, 17521300]. In vitro studies in human hepatocytes have shown the activation of pregnane X receptor (PXR, coded for by NR1I2) and the constitutive androstane receptor (CAR, NR1I3) as the underlying mechanism of the artemisinins-mediated induction of CYP3A4, CYP2B6 and ABCB1 [Articles:15761118, 16919048]. These effects of artemisinin and its derivatives on the above mentioned genes are also important in the content of drug-drug interactions for drugs metabolized by these enzymes, e.g. efavirenz (CYP2B6) [Articles:18223457, 19926036] since many patients receiving these drugs need concurrent treatment for additional infections such as HIV and tuberculosis.
In conclusion, while the candidate genes involved in their production of several metabolites of artemisinins have been identified, further study is needed to explore the impact of genomic variation on these drugs particularly in the context of multiple drug treatments.
Edit history (3)
- 2010-06-12 Update
- 2010-09-28 Update
- 2025-04-10 Update fixed typos