About this pathway
Introduction
The application of surgical treatments is markedly enhanced by the availability of anesthetic agents, such as neuromuscular blockers. Succinylcholine chloride (2,2' - \[(1,4-dioxo-1,4- butanediyl) bis (oxy) ] bis \[ N,N,N-trimethylethanaminium] dichloride), also known as suxamethonium chloride, is a depolarizing neuromuscular blocking agent on the World Health Organization’s List of Essential Medicines. Because of its rapid onset of action and short half-life, it is commonly used in medical procedures that require short-term skeletal muscle paralysis, including rapid intubation in emergency medical situations. The clinical application of succinylcholine (SCH) is tempered by the occurrence of rare, but dramatic adverse reactions and some were the earliest known examples of pharmacogenetics. In many cases, patients with functionally characterized single nucleotide polymorphisms (SNPs) in specific genes in either the pharmacokinetic or the pharmacodynamic pathways of SCH are at increased risk of these adverse reactions. The ubiquity of SCH in medical procedures makes understanding the pharmacogenomics of SCH critical for identifying susceptible patients such that suitable interventions and alternatives may be utilized.
Pharmacodynamics
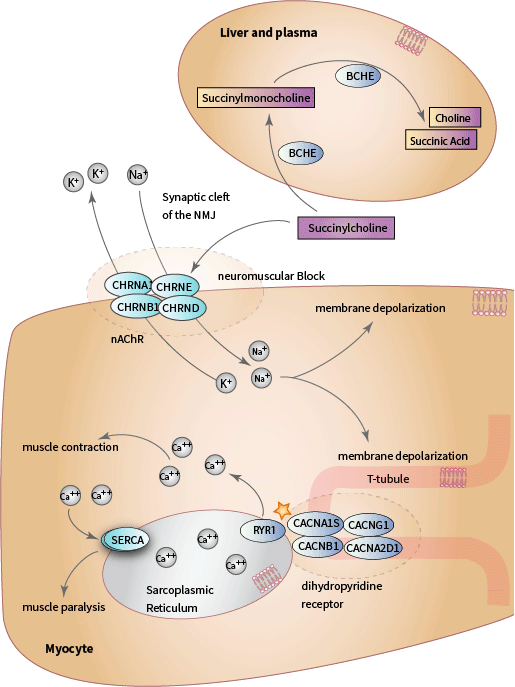
Structurally, SCH consists of two acetylcholine (ACh) molecules linked end to end by their acetyl groups DailyMed Anectine Drug Label and Pharmacology of neuromuscular blocking drugs. ACh is the endogenous agonist of the nicotinic acetylcholine receptor (nAChR), a ligand-gated, nonspecific cation channel that is formed by five subunits organized around a central pore. There are two a 1 subunits, and one ß 1,d , and e subunits. Each subunit of the nACHR is encoded by one of four genes ( a is encoded by CHRNA1, ß 1 is encoded by CHRNB1 , d 1 is encoded by CHRND, and e is encoded by CHRNE) (Fig. 1). The nAChR is located on the motor end plate of neuromuscular junction (NMJ) of the skeletal muscle membrane, also known as the sarcolemma. Binding of an agonist, such as ACh or SCH, promotes the open state of the channel. When the nAChR opens, sodium ions rush into the cell and potassium ions rush out, resulting in membrane depolarization and generation of an action potential. In myocytes, depolarization stimulates muscle contraction [Articles:19417616, 19126755, 24556925].
L-type voltage-gated calcium channels, also known as dihydropyridine receptors (DHPR), are located on invaginations of the sarcolemma called the transverse tubules (T- tubules). The DHPR is a complex of four subunits ( a 1, a 2 d , ß , ¿ ) and a distinct gene encodes each subunit. CACNA1S encodes the a 1 subunit (also called Cav1.1), the primary subunit of the channel that contains the voltage sensor, gating apparatus, and channel pore of DHPR [Article:16382099]. The DHPR is mechanically coupled to the ryanodine receptor (RYR1), a homotetrameric voltage-gated calcium channel that is located on the sarcoplasmic reticulum (SR) and encoded by the RYR1 gene [Article:21798098] (see pathway diagram). When skeletal muscles are at rest, the troponin complex allosterically inhibits the formation of a cross-bridge between myosin and actin. When calcium is released into the myoplasm, it binds the troponin complex and allows myosin to bind to actin to initiate muscle contraction and continues for as long as ATP is freely available [Article:25294644]. Upon depolarization of the sarcolemma, the DHPR undergoes a conformational change and transmits a signal to RYR1, which opens to release SR calcium stores into the myoplasm to initiate muscle contraction. Muscle relaxation occurs when calcium ATPases on the sarcoplasmic reticulum (SERCA) remove calcium from the myoplasm and pump it back into the SR [Article:19789383].
Because it is a depolarizing neuromuscular blocking agent, SCH first induces muscle fasciculations, followed by flaccid muscle paralysis. SCH takes effect within 60 s of intravenous administration and paralysis lasts between 4 and 6 min, during which time patients are monitored with an electric nerve stimulator DailyMed Anectine Drug Label. Because of its short half-life, SCH is indicated for medical procedures requiring short-term muscle paralysis, such as endotracheal intubation, neuromuscular surgery, and electroconvulsive therapy. Because SCH paralyzes the respiratory muscles, patients require mechanical ventilation and close monitoring for the duration of paralysis. It exerts no direct effect on smooth or cardiac muscle contraction. SCH is often administered in combination with other anesthetics, analgesics, and narcotics because although it blocks muscle contraction it has no effect on pain perception DailyMed Anectine Drug Label , Pharmacology of neuromuscular blocking drugs ,and [Article:16402115].
Pharmacokinetics
SCH is rapidly hydrolyzed by butyrylcholinesterase (BCHE, also known as plasma cholinesterase and pseudocholinesterase), which is synthesized in the liver and present in plasma. BCHE hydrolyzes SCH to succinylmonocholine, succinic acid, and choline [Article:2195556] (see pathway diagram). The duration of the neuromuscular block caused by SCH is determined both by its rate of dissociation from the nAChR as well as the rate of metabolism by BCHE in the plasma. ACh is rapidly metabolized by acetylcholinesterase, but unlike acetylcholinesterase, BCHE is not present in the synaptic cleft of the NMJ. Consequently, SCH is hydrolyzed more slowly and remains bound to the nAChR for longer than ACh. For as long as SCH is bound to the nACHR, it maintains the membrane potential above the threshold for repolarization and ACh cannot trigger depolarization of the sarcolemma until it has been repolarized DailyMed Anectine Drug Label.
Pharmacogenomics
Prolonged Apnea
The adoption of SCH as a muscle relaxant was rapid and widespread after it was introduced in the early 1950s, but doctors sometimes encountered patients in whom the paralyzing effects of SCH lasted considerably longer than normal, placing the patients at risk of asphyxiation because of prolonged incapacitation of their respiratory muscles (prolonged apnea). In 1956, Dr Werner Kalow and colleagues reported that the prolonged apnea was caused by the presence of an ‘atypical’ variant (termed the A-variant) of BCHE that impairs its ability to bind and hydrolyze SCH compared with the ‘usual’ variant (termed the U-variant). They also devised a phenotyping assay, still used today, called the ‘dibucaine number’ (DN) test to detect the presence of the A-variant [Articles:2195556, 13479831, 16968950].
BCHE
The BCHE gene is located on chromosome 3q26.1–26.2 and is ~ 64 kb long [Articles:25054547, 21228368]. Since the initial discovery of the A-variant of BCHE, over 60 polymorphisms in the BCHE gene have been reported [Article:21228368]. Genetic variants that impair BCHE enzyme activity can be broadly classified as being either ‘qualitative’ or ‘quantitative’ variants. The qualitative variants affect BCHE enzyme substrate affinity, whereas the quantitative variants affect the quantity of enzyme that is produced without affecting BCHE substrate affinity. Both types of variants increase a patient’s risk of prolonged apnea in the presence of SCH, but the duration of the apnea depends on the type and the number of variants present [Articles:2195556, 21228368] and OMIM. A list of the most common genetic variants of BCHE can be found in Table 1.
Table 1: BCHE polymorphisms associated with prolonged apnea
| rsID | Amino Acid Translation NP_000046.1 | Phenotype | cDNA change/alleles NM_00055.2 | Alternate Names |
|---|---|---|---|---|
| rs28933389 | Thr271Met | Resistant to inhibition by sodium fluoride; decreases BCHE enzyme activity | c.812G>A | F- Variant; BCHE*243M;Thr243Met |
| rs28933390 | Gly418Val | Resistant to inhibition by sodium fluoride; decreases BCHE enzyme activity | c.1253C>A | F-Variant; BCHE*390V; Gly390Val |
| rs1799807 | Asp98Gly | Resistant to inhibition by dibucaine; decreases the affinity of BCHE for SCH | c.293T>C | A-Variant; Che*70G; Asp139Gly; Asp70Gly |
| rs121918558 | Tyr156Cys | Significant decrease in BCHE levels | c.467T>C | Tyr128Cys |
| rs104893684 | Leu335Pro | BCHE is absent from plasma; found only in the Vysya of India | c.1004A>G | S-Variant; Leu335Pro |
| rs121918557 | Leu358Ile | Resistant to inhibition by sodium fluoride and dibucaine; decreases BCHE enzyme activity; found only in Japan | c.1072A>T | F-Variant; Leu330Ile |
| rs121918556 | Glu525Val | Causes a 66% reduction in circulating enzyme and decrease in BCHE activity | c.1574T>A | J-variant; Glu497Val |
| rs1803274 | Ala567Thr | 30% reduction in BCHE enzyme activity | c.1699C>T | K-variant; CHE*539T; Ala539Thr |
| N/A | No change | No change in BCHE activity | None | U-Variant |
Source: http://www.omim.org/entry/177400 and [Article:21228368]
The A-variant (NP_000046.1:p.Asp98Gly; rs1799807 T>C) is the most common qualitative variant, with an incidence of 1 : 3500 in Whites [Article:2195556]. The A-variant results in a BCHE enzyme that is resistant to inhibition by dibucaine and can be detected by the DN test. The DN number indicates the percentage of BCHE enzyme activity remaining after inhibition by dibucaine. Patients with a very low DN are characterized as being homozygous for the A-variant (AA) [Articles:2195556, 21228368, 13437188]. A common quantitative variant (NP_000046.1:p.Ala567Thr; rs1803274 C>T) is the Kalow variant (termed the K-variant). The K-variant decreases BCHE enzyme levels by 30% and is often in linkage disequilibrium with the A-variant [Articles:21228368, 15731589]. Less common variants of BCHE include three fluoride resistant variants (rs28933389, rs28933390, and rs121918557) and many silent variants (termed ‘S-variants’). Some S-variants lead to marked reductions of BCHE (Table 1). Multiple mutations, including point mutations, deletions, insertions, and combinations of mutations, may result in the S-variant form of BCHE, including some that have only been described in specific populations as private variants [Articles:25054547, 16788378].
Although DN indicates whether a patient carries an A-variant, other tests are required to indicate whether a patient is a carrier of an F-variant or an S-variant (tests of fluoride resistance and tests of BCHE enzyme activity, respectively). BCHE is highly polymorphic, and compound homozygosity and heterozygosity have been observed in patients with BCHE deficiency [Article:25054547]. An Australian study reported that in 44% of the BCHE deficient patients who were genotyped, the most common BCHE variant was the compound homozygous A-variant and homozygous K-variant [Article:12881446]. Because the condition is rare, and it can only indicate whether an individual is a carrier of the atypical variant (the A-variant) of BCHE, the DN test is not used routinely to check for the presence of BCHE deficiencies. According to Miller’s Anesthesia (8th ed.), the DN test is indicated for individuals suspected of carrying the A-variant. This is important as this condition can also be caused by environmental factors such as exposure to organophosphate pesticides, liver diseases, and pregnancy [Article:2195556]. In cases of suspected BCHE deficiency caused by environmental factors, enzymatic activity testing, rather than genetic testing, could be useful before administration of SCH (Miller's Anesthesia, 8th edition).
Malignant Hyperthermia
SCH can also trigger malignant hyperthermia (MH) in some patients. MH was first described in Australia in 1960. A patient expressed concern about being under general anesthesia for his surgery because ten of his family members had died while under anesthesia or shortly afterward. The doctor, assuming that ether had been the cause of death, administered halothane instead, but shortly after being administered halothane, the patient experienced a severe decrease in blood pressure and a rapid increase in body temperature. By packing the patient with ice, the doctor stabilized the patient’s body temperature and saved his life. The patient was referred to the geneticist Dr Michael Denborough. Dr Denborough concluded that the patient was a carrier of an autosomal dominant mutation of incomplete penetrance that made him and members of his immediate family susceptible to hyperthermia in the presence of general anesthesia [Article:18156894]. Other case studies emerged of patients who experienced masseter spasm and jaw rigidity in addition to hyperthermia after administration of SCH [Article:21624965].
Malignant hyperthermia susceptibility (MHS) is inherited in an autosomal dominant manner that manifests in a subset of patients who are administered depolarizing muscle relaxants, such as SCH, or potent inhalational anesthetics such as sevoflurane, isoflurane, or desflurane. Epidemiological reports provide the occurrence of MH as ranging anywhere from one in 5000 to one in 100,000 patients; however, as it only reveals itself in the presence of potent inhalational anesthetics and depolarizing muscle relaxants, the genetic prevalence of MHS has been estimated to be as high as one in 2000 [Articles:17456235, 19018722, 24433488, 24868161]. Given that the overall incidence of MH in the general population is low, it is likely that sporadic causative mutations are rare [Article:24445638]. Most MHS-causative mutations are inherited in an autosomal dominant manner, which explains why MHS heritability is so high [Article:16917943].
Multiple epidemiological studies also show that males are overrepresented among surgical patients diagnosed with MH. For example, in two retrospective studies of patients who had experienced MH episodes during anesthesia, between 64.8 and 70% were male [Articles:24433488, 24868161, 24974921]. This could be because of bias in the types of surgeries requiring general anesthesia that may be more commonly performed in men. However, one study found that men were more likely to be diagnosed with MHS than women on the basis of in-vitro contracture test (IVCT, see below) even when there was no statistically significant difference in the numbers of men and women in the samples [Article:17430325].
Early symptoms of MH include metabolic (elevated CO2 production and O2 consumption), cardiovascular (tachycardia and arrhythmia), and muscular abnormalities (masseter spasm and muscle rigidity). Later and more severe symptoms can include a rapid increase in body temperature (hyperthermia), rhabdomyolysis, hyperkalemia, severe metabolic and respiratory acidosis, and cardiac arrest DailyMed Anectine Drug Label and [Articles:19018722, 20837722]. A clinical grading scale is a scoring system used to estimate the likelihood that a patient administered anesthesia has experienced an MH episode on the basis of multiple indicators, such as the aforementioned symptoms [Article:24433488]. The mortality rate for patients who experience an MH crisis has decreased significantly to between 5 and 10% with the introduction of dantrolene, an RYR1 inhibitor; before dantrolene, the mortality rate was as high as 70% [Article:19018722].
MH is characterized by a rapid and uncontrolled increase in myoplasmic calcium levels in skeletal muscle in response to depolarizing muscle relaxants or potent inhalational anesthetics, followed by a hypermetabolic state caused in part by the ATP-intensive removal of excess calcium by SERCA and other calcium pumps [Article:21798098]. Two mechanisms of extracellular calcium entry have been proposed to explain how an MH crisis can be maintained if SR calcium stores are limited. Store-operated calcium entry promotes extracellular entry of calcium into cells when SR calcium stores are depleted and excitation contraction-coupled entry promotes extracellular calcium entry independent of SR calcium stores, but is triggered by prolonged depolarization of the sarcolemma [Articles:17942409, 20566647]. There is evidence that store-operated calcium entry as well as excitation contraction-coupled entry are induced in muscles with RYR1 mutations, but not in wild-type muscles in the presence of low levels of halothane. The identities of all of the calcium channels responsible for extracellular calcium entry are still being discovered [Articles:24411466, 23159934].
The role of SCH as a trigger in MHS individuals is still unclear because although it has been reported to trigger MH when administered alone, the majority of MH crises have occurred when SCH was coadministered with a potent inhalational anesthetic [Articles:21624965, 19018722, 24433488]. One retrospective analysis reported that the relative risk of MH is greater when potent inhalational anesthetics and SCH are co-administered rather than when either is administered alone [Article:23223104]. A more recent analysis concluded that there was no significant difference in the severity of MH when comparing between those who were administered potent inhalational anesthetics alone and those who were co-administered SCH. However, the study did report that the onset of an MH crisis was significantly more rapid in patients who were coadministered SCH and potent inhalational anesthetics compared with either alone [Article:24433488]. A third study reported that SCH was associated with greater elevations of serum creatine kinase levels in MHS patients compared with when SCH was not administered [Article:7486044].
Genetics of malignant hyperthermia
The most commonly reported mutations in MHS individuals are in RYR1. As of October 2013, there were at least 397 reported SNPs in RYR1, but only 33 have been functionally characterized and designated as ‘MHS causative’ by the European Malignant Hyperthermia Group (EMHG), a research consortia specializing in MH Genetics in Malignant Hyperthermia. There are also polymorphisms in CACNA1S, associated with MHS, but 30–50% of patients who are diagnosed with MHS have no known MHS-causative mutations in either CACNA1S or RYR1 [Articles:19018722, 24433488, 24868161, 16917943]. Studies in vitro, in mouse models, as well as in humans have shown an association between mutations in RYR1 and CACNA1S, MHS, and elevated basal levels of myoplasmic calcium in skeletal muscle. Why some mutations in RYR1 or CACNA1S could lead to elevated basal calcium is not yet known, although elevated calcium has been attributed to increased sensitivity of RYR1 to agonists because of increased passive leaks from the SR [Articles:23159934, 22547813, 11524458, 17182726, 15537710]. The American College of Medical Genetics’ 2013 report for recommendations on reporting incidental findings in clinical exome and genome sequencing also includes RYR1 and CACNA1S in their list of genes to report [Article:23788249].
RYR1
RYR1, the primary locus of MHS, is located on chromosome 19q13.1–13.2. RYR1 is found complexed with other proteins, but because most pharmacogenetic studies of MHS are on RYR1, they will not be discussed here. Early efforts to sequence RYR1 were hampered because of the large size of the gene; RYR1 is ~ 16 kb and contains 106 exons [Article:16917943]. There are three recognized ‘hot-spots’ in RYR1 in which SNPs are often found: the N-terminus, C-terminus, and a central region that includes exons 39, 40, and 44–46 [Article:15448513]. The location of individual mutations in RYR1 or CACNA1S is likely to determine the specific mechanism responsible for producing elevations in resting calcium levels, but with so many different MH causative mutations (particularly in RYR1), a single mechanism is not likely to be found. For example, a study in one RYR1 knockin mouse (Tyr522Ser) reported that the mutation was associated with increased RYR1 sensitivity to pharmacologic agonists as well as to stimulation by depolarization, but it had no effect on basal calcium levels [Article:16284304]. A second study in a different RYR1 knockin mouse (Arg163Cys) reported that the mutation was associated with elevated basal calcium levels as well as increased sensitivity to RYR1 agonists compared with the wild-type mouse [Article:17122579].
Several mutations in RYR1 are also associated with var- ious neuromuscular diseases including central core disease (CCD) and multiminicore disease. The clinical presentation of these diseases is variable and they may be distinguished histologically [Articles:16917943, 23424048]. Some individuals may experience hypotonia and delayed motor development from infancy, whereas others may be unaware that they have a neuromuscular disease until they are diagnosed for muscle weakness or tested for MHS well into adulthood [Articles:21798098, 8220423]. Although SCH and potent inhalational anesthetics are contraindicated in patients with CCD, the disease does not always segregate with MHS, nor do MHS causative RYR1 mutations always segregate with CCD, further complicating the relationship between RYR1 and MHS [Articles:23424048, 10618932].
The currently accepted ‘gold standard’ for diagnosis is the IVCT. An almost identical procedure called the caffeine–halothane contracture test (CHCT) is used in the USA [Article:2675676]. The IVCT is an invasive test that requires freshly dissected muscle fibers. Muscle fibers are exposed to caffeine and halothane separately, and the force of contracture is measured using a myoelectrical transducer. An MHS-positive IVCT is when the force of the contracture is greater than 2 mN in the presence of 2mmol/l or less of caffeine and 0.44mmol/l or less of halothane [Articles:24433488, 24868161]. Patients can be diagnosed as MHS (sensitive to both halothane and caffeine) or MH negative (sensitive to neither). Patients who are sensitive to either halothane or caffeine, but not both, are MHSc for caffeine or MHSh for halothane [Article:24433488]. The EMHG has recently revised its guidelines regarding the use of genetic testing to diagnose MHS. Now, IVCT is no longer the primary diagnostic test for MHS and anyone in whom an MHS-causative mutation is detected is now automatically diagnosed as MHS. The EMHG still recommends that MHS individuals and relatives in whom an MHS-causative mutation is not detected should still be tested with IVCT because of the possibility of false negative diagnosis, due in part to the possible presence of novel MHS-causative mutations in RYR1 [Article:26188342]. The EMHG has established guidelines for how to recognize and treat patients during a fulminant or an abortive MH crisis, guidelines for molecular genetic testing, and a protocol for IVCT testing. In the case of a positive MH diagnosis, positive IVCT, or MHS-positive genetic test, the EMHG strongly recommends that IVCT and genetic counseling be offered to first-degree relatives of the proband [Articles:20837722, 26188342]. The Malignant Hyperthermia Association of the United States explains that RYR1 hot spots are sequenced first in an effort to detect the presence of known causative mutations (33 as of the writing of this manuscript) and the remaining exons are sequenced if no causative mutations are found in those hot spots Testing for malignant hyperthermia susceptibility. Discordance between IVCT phenotype and RYR1 genotype may possibly be because of the heterogeneity of the RYR1 gene and complexity of the MHS phenotype [Articles:16917943, 10352931]. A list of the EMHG’s MHS causative mutations in RYR1 can be found in Table 2.
Table 2: RYR1 polymorphisms associated with MHS
| rsID | Amino Acid Translation NP_000046.1 | Phenotype | cDNA change/alleles NM_00055.2 |
|---|---|---|---|
| rs193922747 | Cys35Arg | MHS Causative | c.103T>C |
| rs118192161 | Arg163Cys | MHS Causative; CCD associated | c.487C>T |
| rs193922753 | Arg163Leu | MHS Causative | c.488G>T |
| rs1801086 | Gly248Arg | MHS Causative | c.742G>A; c.742G>C |
| rs121918592 | Gly341Arg | MHS Causative | c.1021G>C; c.1021G>A |
| rs118192116 | Ile403Met | MHS Causative | c.1209C>G |
| rs118192162 | Tyr522Ser | MHS Causative; CCD associated | c.1565A>C |
| rs111888148 | Arg530His | MHS Causative | c.1589G>A |
| rs193922770 | Arg552Trp | MHS Causative | c.1654C>T |
| rs118192172 | Arg614Cys | MHS Causative | c.1840C>T |
| rs193922772 | Arg614Leu | MHS Causative | c.1841G>T |
| rs118192175 | Arg2163Cys | MHS Causative; CCD associated | c.6487C>T |
| rs118192163 | Arg2163His | MHS Causative; CCD associated | c.6488G>A |
| rs118192176 | Val2168Met | MHS Causative | c.6502G>A |
| rs118192177 | Thr2206Arg | MHS Causative | c.6617C>G |
| rs118192177 | Thr2206Met | MHS Causative | c.6617C>T |
| rs112563513 | Arg2336His | MHS Causative | c.7007G>A |
| rs193922802 | Ala2350Thr | MHS Causative | c.7048G>A |
| rs193922807 | Gly2375Ala | MHS Causative | c.7124G>C |
| rs193922809 | Ala2428Thr | MHS Causative | c.7282G>A |
| rs121918593 | Gly2434Arg | MHS Causative | c.7300G>A |
| rs28933396 | Arg2435His | MHS Causative; CCD associated | c.7304G>A |
| rs193922816 | Arg2454Cys | MHS Causative | c.7360C>T |
| rs118192122 | Arg2454His | MHS Causative; CCD associated | c.7361G>A |
| rs28933397 | Arg2458Cys | MHS Causative | c.7372C>T |
| rs121918594 | Arg2458His | MHS Causative | c.7373G>A |
| rs118192178 | Arg2508Cys | MHS Causative | c.7522C>T |
| rs118192167 | Tyr4796Cys | MHS Causative; CCD associated | c.14387A>G |
| rs121918595 | Thr4826Ile | MHS Causative | c.14477C>T |
| rs193922876 | His4833Tyr | MHS Causative | c.14497C>T |
| rs193922878 | Leu4838Val | MHS Causative | c.14512C>G |
| rs63749869 | Arg4861His | MHS Causative; CCD associated | c.14582G>A |
| rs118192170 | Ile4898Thr | MHS Causative; CCD associated | c.14693T>C |
Source: http://www.emhg.org/genetics [Articles:19018722, 11573677]
CACNA1S
CACNA1S, the second gene implicated in MHS, is 93.5kb long and is located on chromosome 1q.32. Currently, there are two missense SNPs in CACNA1S that are designated to be MHS causative by the EMHG (Table 3) [Articles:22547813, 15201141]. Monnier et al. [Article:9199552] carried out a linkage analysis in a large French family after the death of a proband following a suspected MH crisis shortly after he was administered SCH and isoflurane. Ten out of eighteen members of his family were subsequently diagnosed with MHS and three were diagnosed with MHEh by IVCT. After determining the locus that segregated with MHS, the authors carried out sequence analyses and discovered the causative mutation in CACNA1S. The mutation (NP_000060.2:p.Arg1086His; rs1800559 C > T) causes an arginine residue to be substituted by a histidine at position 1086. This polymorphism was also found in a large North American case–control study in two patients diagnosed with MHS by IVCT [Article:11260227]. The second SNP (NP_000060.2: Arg174Trp c.520 C > T; rs772226819) was discovered in a study that sequenced CACNA1S cDNA in 50 MHS-positive individuals in the UK. The mutation was found to segregate with MHS in one family and was not found in any of the samples from MHN patients [Article:19825159]. Polymorphisms at additional loci, including the gene encoding the a 2 d subunit of the DHPR, have been proposed as being MHS causative, but no strong associations between those polymorphisms and MHS have emerged [Articles:9508059, 7951247, 7887423].
Table 3: CACNA1S polymorphisms associated with MHS
| rsID | Amino Acid Translation NP_000060.2 | Phenotype | cDNA change/alleles NM_000069.2 |
|---|---|---|---|
| rs772226819 | Arg174Trp | MHS Causative | c.520G>A |
| rs1800559 | Arg1086His | MHS Causative | c.3257C>T |
Source: http://www.emhg.org/genetics [Articles:19018722, 11573677]
Hyperkalemia
Hyperkalemia, a serum concentration of potassium exceeding 5.5 mmol/l, is a dangerous condition that can lead to arrhythmia and cardiac arrest [Article:21181208]. The FDA-approved drug label for SCH contraindicates it for patients in the acute phase of injury as well as for patients with genetic mutations that cause Duchenne and Becker’s muscular dystrophies because they are at increased risk of hyperkalemia DailyMed Anectine Drug Label and [Article:16394702]. In skeletal muscle, the nAChR is normally only present at the NMJ, but nerve damage and denervation stimulates nAChR gene expression throughout the muscle membrane. Denervation also stimulates the expression of a second nAChR isoform called a7AChR. a7AChR is a homomeric nonspecific cation channel that responds equally well to SCH and choline [Article:9508059]. The simultaneous upregulation of both nAChR isoforms increases a patient’s risk of excessive potassium release and hyperkalemia when triggered by SCH. Patients with Duchenne and Becker’s muscular dystrophies develop significant denervation of their muscles and are at a high risk of experiencing hyperkalemia if they are administered SCH [Articles:23919455, 19762730].
Edit history (3)
- 2021-08-10 Update Added related inhaled anesthesia pathways.
- 2022-09-12 Update Fixed broken link for MHAUS testing information
- 2022-09-29 Update fixed typos