About this pathway
Background
Lamotrigine (LTG) is an antiseizure drug (ASD) which was approved by the US Food and Drug Administration (FDA) in 1994 to treat focal (partial) seizures, primary generalized tonic-clonic seizures, and generalized seizures of Lennox-Gastaut syndrome in both children and adults. It was evaluated for several mood disorders and subsequently also approved for maintenance of bipolar disorder in adults [Articles:2498073, 8112232, 7968065, 9061824, 9596201]. LTG was originally synthesized at Wellcome Laboratories as an antifolate analog, but screening in animal seizure models revealed potent activity and it was developed to improve upon existing antiepileptic drugs for refractory patients with a better safety and drug interaction profile [Article:18001843]. LTG is a 2,5-diamino-triazine ASD which blocks voltage-gated sodium channels (VGSCs) [Articles:7687190, 9249262, 9765515, 19635852, 24096830, 24283699] and voltage-gated calcium channels (VGCCs) [Articles:7687190, 8831112, 9169293, 9778600] both of which may contribute to the antiseizure activity of this drug.
The common adverse effects for LTG are dizziness, diplopia, headache, ataxia, blurred vision, nausea, somnolence, vomiting, and hypersensitive skin rashes including severe reactions such as Stevens-Johnson syndrome (10% rash incidence) [Articles:9429131, 9740111]. Risk of severe cutaneous adverse reactions (Stevens-Johnson syndrome/Toxic Epidermal Necrolysis) due to LTG is related to HLA-B polymorphisms, including HLA-B*15:02 mostly in Asian (Han Chinese) population (p<0.05) [Articles:17509004, 20485159, 22833576, 22500513, 23692434]. Further, one study reported that HLA-B*15:02 allele was present in 33.3% of lamotrigine-induced SJS/TEN cases whereas 9.4% in lamotrigine-tolerant controls (P < 0.05,), however the sample size was small [Article:25428396].
In this article we review the metabolic pathways and mechanism of action of LTG, along with a comprehensive summary of polymorphisms in pathway genes contributing to the inter-individual variability in LTG clearance and response.
Pharmacokinetics
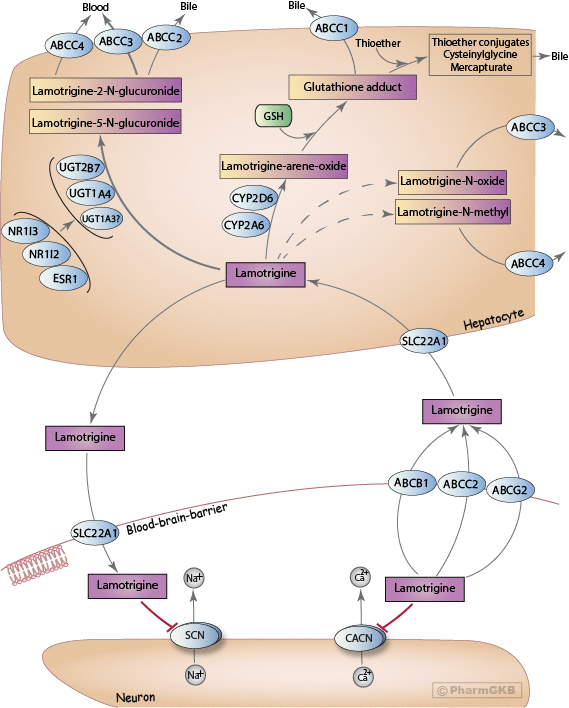
A schematic representation of LTG disposition within the body is shown above. The mechanism by which LTG crosses the blood-brain barrier (BBB) is not completely understood, but studies have implicated a role for organic cation transporters (OCT) [Articles:22227272, 27610747]. An in vitro study showed that LTG is a substrate for SLC22A1 (also known as OCT1) [Article:22227272], and polymorphisms in the SLC22A1 gene have been associated with concentration differences in Chinese patients with epilepsy [Article:27610747].
Although controversial, a current hypothesis for pharmaco-resistant epilepsy implicates overexpression of efflux transporters at the BBB [Article:22197850]. There is conflicting evidence for the role of the ATP-binding cassette (ABC) superfamily of transporters in LTG efflux, specifically P-glycoprotein (P-gp), encoded by the ABCB1 (MDR1) gene [Articles:18824002, 18835265, 22197850] and ABCG2 (BCRP), both located on the apical capillary endothelial membrane [Articles:16529607, 18835265, 23588114, 25645391, 27610747]. Romermann and colleagues recently reported that LTG showed a ABCG2 mediated transport, where LTG exhibited a significant difference in the absence and presence of ABCG2 inhibition (P < 0.0001) [Article:25645391]. Mechanistic study of Löscher and others reported that LTG was an efficient BCRP (ABCG2) substrate in transfected MDCK cells [Articles:25645391, 21827408]. A comprehensive literature review concluded that LTG was a P-gp substrate based on a combination of data from in vitro, in vivo, and predicted structure-activity relationship studies [Articles:18824002, 21827408, 22197850]. Therefore this drug is the first ASD which was identified as a dual substrate of the two major human efflux transporters at the BBB (synergistic or cooperative role of P-gp and ABCG2 in the efflux of dual substrates at the BBB) [Article:25645391]. In contrast, Nakanishi et al. reported no difference in total brain-to-plasma concentration ratios of LTG in Mdr1a/1b/Bcrp triple-knockout mice [Article:23588114]. Despite inconsistencies in model system results, correlations have been identified between polymorphisms in these genes and LTG concentrations in plasma; thus, transporters may play an important role for differential drug response [Articles:16355305, 16857572, 18408562, 19453704, 22972536, 26213157]. Additionally, polymorphisms in ABCC2 (MRP2) are associated with drug resistance in different populations, which indicates this transporter may also play a role in drug efflux [Articles:19415824, 22630058, 27816260].
ABCC3 (MRP3) is located on the sinusoidal membrane of hepatocytes separating the cytosol from the bloodstream, and transports glucuronides from the liver cells into the bloodstream for elimination by the kidney [Articles:16472997, 21103974]. Since 80% of LTG glucuronides are eliminated in urine and the 2-N quaternary ammonium glucuronide is not able to cross membranes, ABCC3 or ABCC4 (MRP4) are likely responsible for transport from the liver to the blood [Articles:11076395, 16472997, 21103974, 24346835]. In contrast, ABCC2 (MRP2) effluxes glutathione adducts from the liver cell out to the bile [Articles:16820223, 18786560]. Glucuronides may also be excreted by ABCC2 into bile, but the major route of excretion is via the urine. Given the ongoing disagreement in results, further research is needed for complete understanding of the actual role of efflux transporters in ASD treatment.
Previous studies mentioned that LTG is extensively metabolized, with over 80% of the total dose recovered in the urine [Articles:3677542, 2804994, 1680630]. Three main metabolites of LTG have been observed in humans: LTG-2-N-glucuronide, LTG-5-N-glucuronide, and a minor LTG-N-oxide [Articles:1795036, 1673389, 17409271, 19387891]. Another minor metabolite, LTG-N-methyl, was also detected in human urine and in dogs but is a minor metabolite in human urine [Article:1795036]. LTG-2-N-glucuronide is the major metabolite eliminated from the body with some 5-N-glucuronide, making up 80-90% of the drug recovered in urine; N-oxide was also detected in patients taking LTG along with the parent drug [Articles:3677542, 1795036, 11087428]. LTG-2-N-glucuronide is an inactive metabolite primarily formed by UDP-glucuronosyltransferase (UGT) 1A4 (UGT1A4) [Articles:1673389, 16565174, 19387891, 19845433]. Additional studies suggest that UGT1A3 (minor compared to 1A4) and UGT2B7 (based on in vitro inhibition with zidovudine [Article:16565174]) may also play a role in the metabolism of LTG, though contradictory evidence exists because glucuronidation was not confirmed with cloned, expressed UGT2B7 [Article:19387891]. However, there are studies reported that polymorphisms (such as UGT2B7_-161C>T, UGT2B7 372 GG) in UGT2B7 have been associated (p< 0.05) with LTG concentration-to-dose ratio and clearance which suggests a possible influence of UGT2B7 in LTG metabolism [Articles:20216122, 23263737, 27096250, 29395496].
Previous studies also reported about minor pathway of LTG bioactivation to a reactive arene oxide on the dichlorophenyl ring [Articles:11087428, 19961160]. The P450 isoenzymes mainly responsible for the formation of the arene oxide are CYP2A6 and CYP2D6 [Articles:11087428, 17409271, 19961160]. Chen et al. identified a glutathione conjugate in human and rat liver microsomal incubations in the presence of NADPH and glutathione (GSH) [Article:19961160]. Four radiolabeled metabolites were identified and quantified radiometrically in rat bile (biliary metabolites) by HPLC; of these four, the most polar metabolite was the protonated molecule of a glutathione adduct of LTG (i.e. the primary thioether addition product of an arene-oxide). Glutathione-derived adducts produced thioether conjugates, such as a cysteinyl-glycine adduct of LTG and a cysteine adduct of LTG. [Articles:11087428, 19961160]. However, glutathione-derived N-acetylcysteine conjugates (mercapturic acids) have not been measured in human urine [Articles:11087428, 19961160]. The enzyme likely responsible for formation of mercapturic acids from cysteine S-conjugates has been identified as NAT8 [Article:20392701]. Formation of reactive metabolites (formed from the arene oxide) has also been observed in keratinocytes and may be responsible for mild skin rashes and more severe cutaneous reactions observed with LTG administration [Articles:18786560, 19961160].
One study reported that concomitant administration with UGT-inducing antiseizure drugs such as phenytoin, phenobarbital, or carbamazepine, the elimination half-life of LTG decreases by ~40-50% [Article:3504397]. In contrast, another study reported that valproic acid (VPA) inhibits LTG metabolism, increasing half-life linearly (from 24 h to ~72 h) with increasing dose [Article:11128232]. Addition of VPA may counteract the change in kinetics caused by co-administered UGT-inducing antiseizure drugs [Article:3504397]. In vitro studies with VPA have shown the increased area under the LTG plasma-concentration time curve to be due to competitive inhibition of UGT1A4 or UGT2B7 by VPA. [Article:16565174]. VPA has higher affinity for UGT2B7, but it is also a substrate of UGT1A4, albeit with a higher Km of 3.1 mM, compared to 1.2 mM for UGT2B7 [Articles:18838507, 19387891]. Thus, the LTG-VPA interaction may be mediated by competitive binding to these two UGT isoforms.
Decreased LTG plasma concentrations caused by an increase in drug clearance have been reported during pregnancy [Articles:9579945, 12136066, 15304599, 14745072, 29588992]. A population pharmacokinetic study identified two subpopulations of women who experience different changes in LTG clearance during pregnancy, which may indicate that a genetic mutation is responsible for these differences [Articles:24883336, 29395496]. It is hypothesized that the increase in 17β-estradiol during pregnancy results in activation of estrogen-receptor-α (encoded by the ESR1 gene) in the liver, resulting in upregulation of UGT1A4 expression and increased LTG clearance [Article:19546240]. However, evidence for direct binding of estradiol to estrogen response elements in the UGT1A4 promoter is lacking, but an indirect effect of ERα on the SP1 transcription factor (specificity protein-1) is more likely responsible for the induction observed during pregnancy [Article:19546240]. Furthermore, many studies showed similar increases in LTG clearance occurs with concomitant use of oral contraceptives containing estrogen [Articles:12939444, 17346247, 18201913], but not in progesterone-only based contraceptives [Article:16146436]. An additional piece of evidence includes the observation that UGT1A4 & UGT2B7 expression correlate with ESR1 expression [Articles:24879639, 27899892].
Expression of genes in the UGT1A and UGT2B families are also induced by activators of two nuclear receptors pregnane-X-receptor (NR1I2, also known as PXR), and constitutive androstane receptor (NR1I3, also known as CAR) [Articles:22371261, 24879639, 30253071]. Many studies showed that, UGT1A4 specifically has been shown to be co-regulated by CAR, PXR, and aryl hydrocarbon receptor (AhR) [Articles:24641623, 27899892, 29610665, 29440451, 30253071], while UGT2B7 is co-regulated by CAR and PXR [Articles:21415305, 24879639, 27899892, 30253071]. Carbamazepine, phenytoin, and phenobarbital are prototypical CAR activators e.g. leading to induction of CYP2B6, and these drugs increase LTG clearance when co-administered for epileptic seizures [Article:19845433].
Pharmacodynamics
A stylized depiction of the potential mechanism of action of LTG is shown above. The exact mechanism through which LTG elicits its therapeutic effect is unknown; however, a likely mechanism is through antagonizing type 2 voltage-gated sodium channels (VGSCs; encoded by the SCN gene family) [Articles:7687190, 24096830, 24283699], similar to the mechanism of the older antiseizure drugs phenytoin and carbamazepine [Articles:15638774, 24283699]. VGSCs are large membrane-spanning proteins consisting of a large alpha subunit, which sometimes interacts with a smaller regulatory beta subunit. At resting potential, the ion pore exists in the closed state, but once neuronal membranes containing VGSCs are sufficiently depolarized (typically by an action potential), there is a structural change in the protein causing the ion pore to open. Within milliseconds, the inactivation loop moves to block the flow of ions through the pore, during which point the channel is said to be inactivated. LTG preferentially binds to the inactivated state of VGSCs, acting as an antagonist [Articles:8394919, 7491269, 9249262]. The binding site for LTG is located on the extracellular side of the alpha subunit and is shared by carbamazepine and phenytoin [Article:9765515].
LTG also acts as an antagonist to high-voltage-activated N-, P-, and Q-type calcium channels (VGCCs; encoded by the CACN gene family) [Articles:7687190, 8831112, 9579933, 9169293, 9778600], which may be an additional mechanism through which LTG elicits its antiseizure properties. VGCCs are structurally similar to VGSCs, however they do not have the intracellular inactivation loop present in VGSCs. As such, VGCCs can only exist in an open and closed state, and they are normally closed at the resting membrane potential [Articles:17126810, 21660289]. The blockade of voltage-gated sodium and calcium channels modulate the release of neurotransmitters. Studies consistently show a reduction in release of the excitatory neurotransmitter, glutamate, upon LTG administration [Articles:3757936, 8570024, 7477991, 8923665, 10188980, 10963757]. However, LTG has been reported to elicit both increased [Articles:10963757, 11839834] and decreased [Articles:3757936, 7687190, 8570024, 7477991] release of the inhibitory neurotransmitter GABA. The role of calcium as a second messenger within the cell may also lead to multifactorial changes as a result of altered ion flux. For example, an in vitro study using primary mouse neuronal cultures found CaM kinase II activity to be affected as a result of LTG administration, leading to altered intracellular calcium concentrations [Article:23227957]. Further studies may fully elucidate the downstream effects of LTG administration and their role in the observed antiseizure activity.
Pharmacogenomics
Metabolizing enzyme variants
Inter-individual variability in clinical efficacy or adverse effects following treatment with LTG has been shown to be associated with genetic variants within drug metabolizing enzymes, drug transporters, and drug targets [Articles:21557672, 23263737, 24820767]. As LTG is metabolized by UGTs, single-nucleotide polymorphisms (SNPs) in UGT1A4 and UGT2B7 may play a role in the inter-individual variability in LTG metabolism [Articles:20692811, 21557672]. Two promoter polymorphisms in UGT1A4, -219C>T (rs3732219) and -163G>A (rs3732218), have been associated with altered LTG pharmacokinetics [Article:25922177]. Several studies reported that these two promoter SNPs in the 5’-untranslated region of UGT1A4 (rs3732219, rs3732218) are in high linkage disequilibrium with rs2011425 (Leu48Val) with an MAF of 0.082 in Europeans, 0.226 in E. Asians and 0.102 in Africans (gnomAD, dbSNP) [Articles:15855727, 19890225, 25922177]. Further, studies showed that these polymorphisms are associated with lower enzymatic activity (p<0.05) as well as significantly higher LTG concentration (p<0.01) as compared to wild-type [Articles:23371966, 25922177]. Mechanistic study showed that, the two UGT1A4 promoter polymorphisms (rs3732218 -163G>A allele and rs3732219 -219C>T allele) have been associated with a reduction in basal UGT1A4 luciferase reporter activity by 40-50% in MCF7 breast cancer cells and 30-40% in HepG2 hepatoma cells. [Articles:23371966, 24298433]. Additionally, UGT1A4 142T>G (rs2011425) in the coding region was significantly associated with increased LTG concentrations, lower LTG clearance, and better efficacy in treating epilepsy for patients with the TT genotype compared to GT and GG genotypes [Articles:21601426, 22047493, 24820767, 26303110, 25492569]. Further, in vitro studies with cloned, expressed UGT1A4 variant enzymes with either the 142 A>G (Leu48Val, UGT1A4*3) or the Pro24Thr (UGT1A4*2) mutation had approximately 50% reduced intrinsic clearance (Vmax/Km) compared to the wild-type enzyme [Article:22047493]. Several studies reported that pregnancy increases LTG clearance by >50% and UGT1A4 rs2011425 (UGT1A4*3) was associated with reductions in the LTG concentration-to-dose ratio (C/D ratio) during pregnancy [Articles:12136066, 19546240, 29395496]. The rs2011425 polymorphism also showed significant effects on efficacy in pediatric epilepsy patients treated with LTG [Article:27795544]. Mechanistic study described that, individuals who are homozygous for the rs2011425 GG genotype show higher glucuronidation activity compared to individuals with the TT genotype using human liver microsomes isolated from 80 genotyped livers [Articles:15057901, 24641623].The minor allele frequency of rs2011425 is 0.082 in Europeans, 0.101 in Africans, and 0.213 in E. Asians (gnomAD, dbSNP). Saeki et al. defined the UGT1A4*3a haplotype (containing rs3732219, rs3732218, rs2011425 together with the synonymous SNPs 448T>C (L150L), 804G>A (P268P) and IVS1+43C>T) with a frequency of 12.5%, whereas the rare UGT1A4*7a haplotype (MAF = 0.002) is comprised of rs3732219, rs3732218, rs2011425 plus 448T>C, 804G>A and 1VS1+43C>T together with the non-synonymous 271C>T (R91C) [Articles:15708967, 15855727]. Although information on SNPs and haplotypes of UGT1A4 is available at https://www.pharmacogenomics.pha.ulaval.ca/ugt-alleles-nomenclature/. Unfortunately, the impact of the different haplotypes on LTG pharmacokinetics has not been comprehensively investigated. UGT1A4 70C>A (Pro24Thr, rs6755571), located at the end of the signal peptide with an MAF of 0.052 in Europeans, 0.000 in E. Asians, and 0.015 in Africans (gnomAD, dbSNP), was associated with higher LTG serum concentrations and lower clearance in vitro even during pregnancy [Articles:22047493, 24820767, 25492569, 29395496]. The rare UGT1A4 1091C>T SNP (rs34946978, Pro365Leu) located in the UDPGA binding region of all UGT1A isoforms was associated with a general reduction in glucuronidation activity of the entire UGT1A family (MAF = 0.011 in E. Asian, <0.0002 in Africans and Europeans) [Articles:15304120, 21726413].
Contradictory evidence exists for a role for UGT2B7 in the formation of the major metabolite LTG-2-N-glucuronide [Article:16565174]. No activity with commercial cloned, expressed UGT2B7 in Supersomes® was found (RP Remmel, personal communication). Polymorphisms in this gene may play a role in inter-individual variability in LTG concentrations and dose in patients with epilepsy [Articles:20692811, 21557672]. In a study (n = 53), the SNP -161C>T (rs7668258) SNP in UGT2B7 was found to be significantly associated with lower LTG concentration-to-dose ratios in epilepsy patients with the TT genotype compared to patients with the CC genotype [Article:20216122]. These findings were corroborated by another small study from Thailand (n = 75) that found that the TT and CT genotypes had on average 18% lower clearance than patients carrying the CC genotype [Article:23263737]. Patients with epilepsy on stable dosing with LTG with the UGT2B7 -161G>T (rs7668258) TT genotype had a reduced clearance compared with the GT and GG genotypes. Clearance was 247% higher in Slovenian epilepsy patients with the UGT2B7 -372A>G GG genotype as compared to the AA genotype [Article:27096250]. Furthermore, a case study of a 38-year-old woman treated with LTG suggests that the UGT2B7 -372A>G polymorphism may have a role in body rash and multi-organ failure [Article:26173940]. A recent study reported that UGT2B7 802C>T (rs7439366) was associated with reductions of LTG concentration-to-dose ratio (C/D ratio) during pregnancy [Article:29395496].
Transporter variants
ATP-binding cassette efflux transporters are overexpressed at the BBB, where they reduce the penetration of ASDs into the brain. ABCB1 and ABCG2 have been implicated to play a key role in LTG transport. This would suggest that genetic variants of drug transporter genes associated with functional variations in efflux activity may contribute to the inter-individual variation in ASD drug resistance [Articles:12686700, 16355305, 18627414, 20019695, 22972536].
Polymorphisms in transporter proteins have indeed been shown to significantly influence pharmacokinetics and bioavailability of many drugs. There is several evidence that ABCB1 1236 C>T (rs1128503), 2677 G>T/A (rs2032582), and the synonymous, high frequency 3435 C>T SNP (rs1045642) influence LTG serum concentration (1236C-2677G-3435C carriers had higher LTG concentrations than 1236T-2677G-3435T carriers followed by 1236T-2677T-3435C carriers) and drug response in patients [Articles:16355305, 18408562, 22972536, 26213157, 28165634]. Polymorphisms in ABCG2 (rs2231142 and rs3114020) were found to be significantly associated with LTG concentration dose-normalized by body weight [Articles:25645391, 26213157]. A patient’s cohort study with 131 LTG monotherapy patients reported that presence of ABCG2 421C>A (rs2231142) resulted in modestly lower LTG trough concentration (CI-95%) compared to wild type [Article:29791014]. Further, ABCG2 rs2231142 was also shown to be responsible for approximately 4.8% of the variability in LTG trough concentration variation in Chinese epilepsy patients [Articles:26213157, 27610747]. Additionally, ABCC2 -24C>T (rs717620) polymorphism in exon 1 was reported to be associated with resistance to LTG (P<0.001) and other ASDs (VPA, phenobarbital, carbamazepine, and oxcarbazepine) in a German-Caucasian as well as an Asian patient cohort, hypothesized to be a result of compensatory upregulation of ABCB1 [Articles:19415824, 22630058]. Further, linkage disequilibrium (LD) test showed that the ABCC2 rs717620 were in strong LD with rs2273697 (D'= 0.694) and rs3740066 (D'= 0.699) and frequencies of haplotypes (ABCC2 -24C>T/ABCC2 1249G>A/ABCC2 3972C>T) in resistant patients was significantly higher (P < 0.05) in Chinese epilepsy patients (n=537) [Article:22630058] .However, ABCC2 -24C>T was not associated with drug resistance in Han Chinese, Croatian, or Austrian-Caucasian epilepsy patients [Articles:21449672, 22256867, 23697249].
Opposing the force of efflux transporters are influx transporters that act to carry LTG across the BBB into the brain. One such transporter is SLC22A1. A study of Chinese epilepsy patient’s cohort (n= 112) found an association between plasma concentrations of LTG and SLC22A1 1A>G (rs628031) genotype, GG genotype having significantly lower LTG dose-normalized concentrations in plasma (P<0.05) which indicated that polymorphisms in the SLC22A1 gene may have association with LTG concentration differences [Article:27610747].
Pharmacodynamic variants
The primary hypothesis for the antiseizure effect of LTG is through binding to voltage-gated sodium channels [Articles:7687190, 24096830, 24283699]. Voltage-gated sodium channels are heteromeric complexes that regulate sodium exchange between intracellular and extracellular spaces. The alpha subunits are encoded by the SCN gene, of which there are four predominant isoforms in the human brain: SCN1A, SCN2A, SCN3A, and SCN8A. A polymorphism in SCN1A, 1G>A (rs3812718), was significantly associated with effective dose of LTG. Peak plasma concentrations corresponded to effective doses were almost half for wild type compared to variant [Article:20037572]. At the time of writing, polymorphisms in the SCN gene and their influence on ASDs including LTG had not been not well investigated.
Although not thought to be a direct target of LTG, GABA receptors are the principal inhibitory receptor in the central nervous system (CNS), and heterogeneity in the ionotropic GABAA receptor is associated with epilepsy [Article:18946538]. Several antiseizure drugs like barbiturates and benzodiazepine-like agents bind to GABAA and cause alteration in receptor subunits to regulate drug response [Article:15208697]. Polymorphisms of the genes encoding different subunits of GABAA receptors may be associated with ASD response and resistance. GABRA1 rs6883877, GABRA1 rs1157122, GABRA1 rs6892782, GABRA1 rs10068980, GABRA2 rs511310, GABRA3 rs4828696, and GABRA3 rs1112122 were all found to be significantly associated with resistance to LTG, along with other ASDs such as carbamazepine, phenytoin, and VPA [Article:24236484]. However, due to a lack of studies, the influence of variations in GABA receptors is not well understood, therefore they are not represented in diagram.
Variants associated with adverse effects
Drug-induced skin injuries, Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug-induced hypersensitivity syndrome (DIHS) are all reported LTG-related adverse events [Articles:22583732, 22930254, 23084805, 24630038, 25933949, 26538961, 26221907, 25645637, 29027197]. SJS/TEN in LTG-treated pediatric patients (n=486) was reported in many studies and 97% of cases occurred within 8 weeks of initiation of LTG therapy, with a median time to onset of 15 days [Article:29637101]. Human leukocyte antigen (HLA) genes produce proteins which identify foreign particles in the immune system [Article:518865]. The complex of drug antigen/metabolite-human leukocyte antigen (HLA)-T cell receptor (TCR) initiates immune reactions for SJS/TEN. Specific HLA alleles or variant predisposition and interaction with a drug allow the presentation of drug-bound HLA to the TCR, which further triggers the activation of CD8+ cytotoxic lymphocytes and a series of specific immune reactions which ultimately cause keratinocyte apoptosis and severe drug-induced skin injuries [Articles:24897291, 25938070, 26254052, 26254050, 28017537]. HLA-B genotypes and LTG-induced cutaneous adverse drug reaction (LTG-cADR) associations have been described in several reports. HLA-B*15:02 is associated with SJS in response to LTG administration in multiple populations [Articles:17509004, 20485159, 22833576, 22500513, 23692434, 25428396]. Another study reported that HLA-B*38:01 was significantly associated with LTG-related SJS/TEN in a Spanish Caucasian population (p<0.001) [Article:27888155]. HLA-DRB1*04:05 and HLA-DQB1*04:01 alleles occurred in a higher frequency in Japanese patients with LTG-cADRs. These two alleles are in linkage disequilibrium in this population, along with HLA-DQA1*03:03, which is also associated with LTG-cADRs [Article:25845749]. Studies in Korean populations assume a relationship between HLA-B*44:03 and LTG-induced SJS/TEN (odds ratio: 12.75; CI 1.03-157.14; p=0.053), however the number of patients for these study was very low (n=9, n=5 respectively) [Articles:25327504, 26632391] and further detailed study is need to explore the role of HLA-B*44:03 in LTG induced SJS/TEN. HLA-A*24:02 allele and LTG-induced maculopapular eruptions (MPE) (OR 3.949, p=0.005) [Articles:26282450, 28476759]. Additionally, HLA-A*24:02 showed significant association with DRESS (drug reactions with eosinophilia and systemic symptoms), in a Spanish Caucasian population (p<0.001, n=12) [Article:27888155]. Furthermore, a recent study reported HLA-A*24:02 was also associated significantly with SJS induced by LTG in a southern Han Chinese population (p = 0.005, n=91) [Article:28476759]. Two other novel SNPs, rs12668095 near CRAMP1L/TMEM204 and rs79007183 near TNS3, were associated (P=4.89×10(-7), P=3.15×10(-10) respectively) with LTG-induced MPE in a Korean population (n=34 discovery cohort and n=59 validation cohort) [Article:26220383]. HLA-A*02:01:01/HLA-B*35:01:01/HLA-C*04:01:01 haplotypes were also associated with LTG-induced MPE in a Mexican Mestizo population (p<0.0001, though n=21) [Article:25495410]. A recent study in Thai patients reported that HLA-A∗02:07 and HLA-B∗15:02 allele carriers were significantly higher in the LTG-induced skin injuries group than in tolerant controls. Additionally, HLA-A*33:03, HLA-B*15:02, and HLA-B*44:03 were significantly higher in the LTG-induced MPE group (though the study only included a small group of patients), and the authors note that these alleles could be useful screening markers for preventing drug-induced skin injuries before LTG treatment in Thai patients [Article:29238301]. Another recent study found that frequency of the HLA-A*31:01 allele was significantly higher (p<0.001, n>50) in the LTG-induced SCAR (severe cutaneous adverse reactions) group compared to the LTG-tolerant group in Korean population. Therefore, HLA-A*31:01 might be a risk allele for LTG-induced SCAR in Korean population [Article:28351624].
Conclusions
There are several pharmacogenomic studies that reveal important aspects of pharmacokinetics, pharmacodynamics, and mode of action of LTG; however, complete understanding of PK/PD and mechanism of action of LTG through further research is still necessary to improve its therapeutic efficacy. The primary motive for utilizing pharmacogenetics in administration of LTG lies in the ability to understand the influence of genetic polymorphisms in the pharmacokinetics and pharmacodynamics of the drug being metabolized. This will allow physicians to optimize therapeutic doses for patients to provide maximum efficacy and optimal seizure control. We have outlined likely gene candidates, but further studies need to be done to identify a specific set of clinically relevant gene signatures.
Reactions & interactions (50)
-
Activation
ESR1 → UGT1A4
-
Activation
ESR1 → UGT2B7
-
Activation
NR1I3 → UGT2B7
-
Activation
ESR1 → UGT1A3
-
Activation
NR1I3 → UGT1A4
-
Activation
NR1I3 → UGT1A3
-
Activation
NR1I2 → UGT2B7
-
Activation
NR1I2 → UGT1A4
-
Activation
NR1I2 → UGT1A3
-
Biochemical Reaction
lamotrigine → lamotrigine 2-n-glucuronide
-
Biochemical Reaction
lamotrigine → N2-methyl lamotrigine
-
Biochemical Reaction
lamotrigine → lamotrigine-arene-oxide
-
Biochemical Reaction
lamotrigine → lamotrigine n2-oxide
-
Biochemical Reaction
lamotrigine-arene-oxide → lamotrigine glutathione adduct
-
Biochemical Reaction
lamotrigine → lamotrigine 5-n-glucuronide
-
Catalysis
UGT2B7 → Biochemical Reaction
-
Catalysis
UGT1A4 → Biochemical Reaction
-
Catalysis
UGT1A3 → Biochemical Reaction
-
Catalysis
SLC22A1 → Transport
-
Catalysis
ABCC1 → Transport
-
Catalysis
ABCC3 → Transport
-
Catalysis
ABCC4 → Transport
-
Catalysis
ABCC4 → Transport
-
Catalysis
ABCC3 → Transport
-
Catalysis
SLC22A1 → Transport
-
Catalysis
ABCC2 → Transport
-
Catalysis
ABCC4 → Transport
-
Catalysis
ABCC3 → Transport
-
Catalysis
CYP2A6 → Biochemical Reaction
-
Catalysis
CYP2D6 → Biochemical Reaction
-
Catalysis
ABCC4 → Transport
-
Catalysis
ABCC2 → Transport
-
Catalysis
ABCC3 → Transport
-
Catalysis
ABCB1 → Transport
-
Catalysis
ABCC2 → Transport
-
Catalysis
ABCG2 → Transport
-
Catalysis
UGT1A3 → Biochemical Reaction
-
Catalysis
UGT1A4 → Biochemical Reaction
-
Catalysis
UGT2B7 → Biochemical Reaction
-
Inhibition
lamotrigine → SCN
-
Inhibition
lamotrigine → CACNA1
-
Transport
lamotrigine → lamotrigine
-
Transport
lamotrigine glutathione adduct → lamotrigine glutathione adduct
-
Transport
N2-methyl lamotrigine → N2-methyl lamotrigine
-
Transport
lamotrigine n2-oxide → lamotrigine n2-oxide
-
Transport
lamotrigine → lamotrigine
-
Transport
lamotrigine 2-n-glucuronide → lamotrigine 2-n-glucuronide
-
Transport
lamotrigine → lamotrigine
-
Transport
lamotrigine 5-n-glucuronide → lamotrigine 5-n-glucuronide
-
Transport
lamotrigine → lamotrigine
Edit history (5)
- 2019-08-14 Create
- 2020-01-06 Update Edited text to reflect changes made to submitted manuscript
- 2020-03-23 Update Added link to pathway publication
- 2021-01-27 Update Added missing word to PK section
- 2022-05-26 Update Added tag for Neurological agents