About this pathway
Background
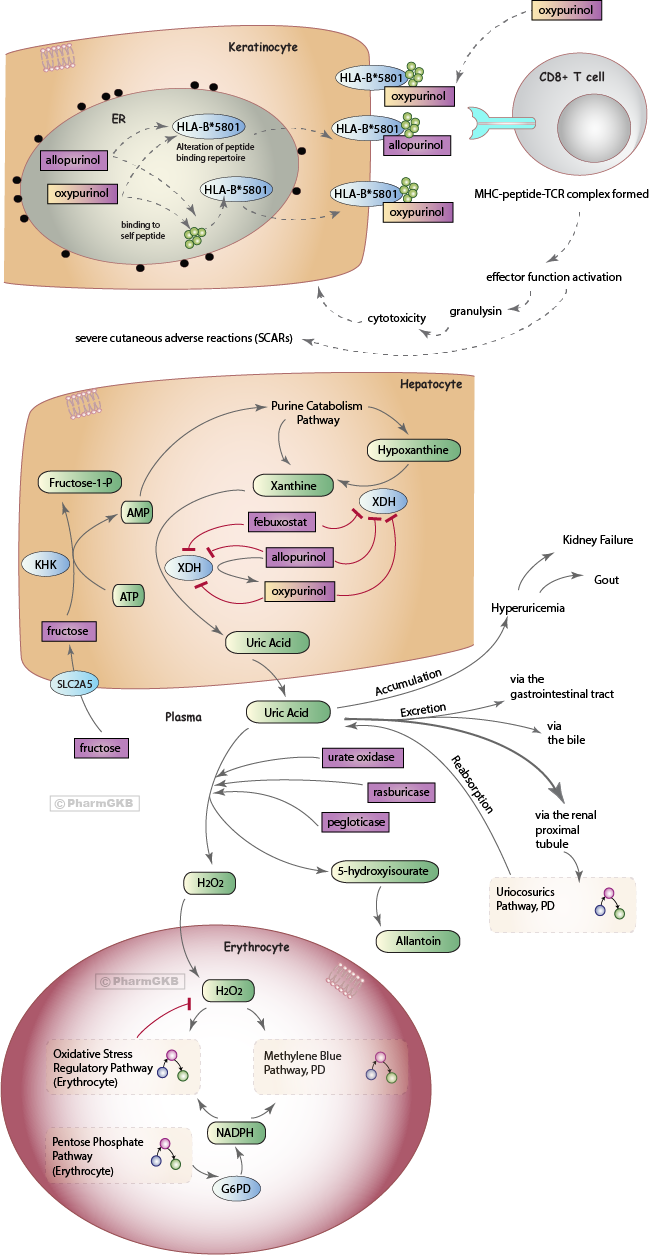
Uric Acid production in humans
Purines are essential for cell growth and survival; as components of DNA and RNA, coenzymes for metabolic reactions, and for cell signaling pathways [Article:11934348]. In humans and great apes, uric acid is the final product of the purine catabolism pathway; lower organisms express urate oxidase which further degrades uric acid, however due to nonsense mutations in the urate oxidase gene (UOX pseudogene) it is not expressed in humans [Articles:12646938, 1765273, 11934348] (Keilin 1959). At physiological pH, uric acid is found predominantly in the form of urate anion [Article:20613716]. Here we encompass urate under the umbrella term 'uric acid'. Uric acid is cleared primarily via the kidneys (>70%), with a smaller proportion via biliary and intestinal secretion [Articles:11934348, 22665944, 22359229, 20613716, 22038265]. However, around 90-95% of uric acid is reabsorbed by transporters in the proximal renal tubule [Articles:20613716, 22359229]. Concentration of plasma uric acid thus depends upon several factors; uric acid generation and excretion, purine de novo synthesis, catabolism and turnover [Article:11934348].
Hyperuricemia
Levels of plasma uric acid above around 7-8mg/dL define 'hyperuricemia'; a condition that results from a higher production or release of uric acid into the plasma exceeding the capacity of normal renal clearance, or situations of reduced clearance or enhanced reabsorption of uric acid [Articles:16597166, 18838473, 22359229] (Fojo, 2011). One of the major risks of hyperuricemia is the development of renal failure due to the crystallization of uric acid in the acidic conditions of renal tubules [Article:12646938]. Chronic kidney disease is therefore often associated with hyperuricemia [Articles:20562597, 12646938, 22665944]. Persistent hyperuricemia can lead to gout (or gouty arthritis) when monosodium urate crystals formed in the joints are released into the joint space and cause inflammation. Gout is a debilitating disease that is coupled with a greater likelihood of co-morbidities and higher mortality [Articles:20562597, 22069122, 22198943, 22945592]. However, since not all patients with uncomplicated hyperuricemia go on to develop gout, drug treatment is not recommended [Article:17349440]. High uric acid levels are also associated with hypertension, obesity and the development of insulin resistance [Articles:19151107, 22359229]. Hyperuricemia is commonly observed in cancer patients and those undergoing chemotherapy, due to higher purine production by malignant cells and release of cell components through increased cell turnover or chemotherapy-induced cell lysis (also known as Tumor Lysis Syndrome, TLS) (Fojo, 2011) [Articles:12646938, 11934348, 18838473].
(non-PMID Reference: Fojo AT: Metabolic Emergencies. In DeVita, Hellman, and Rosenberg's Cancer: Principles & Practice of Oncology, 9th Edition. Volume 9th Edition. Edited by DeVita VTL, Theodore S.; Rosenberg, Steven A.: Lippincott Williams & Wilkins; 2011).
Diet
Diet can contribute to plasma uric acid levels. Alcohol and dietary purines (meat, seafood) are probable risk factors for gout, historically seen as a disease of the rich [Articles:22645377, 23253231, 23175570]. A high guanosine content is found in beer and ethanol increases ATP degradation [Articles:23253231, 23175570]. Sugar (sucrose) is a disaccharide made up of glucose and fructose [Article:23175570]. When glucose is metabolized, intracellular ATP levels are maintained when glucose is metabolized, however; this is not the case with fructose [Article:23175570]. Fructose is transported into cells by SLC2A5 (Glut5) and is metabolized to fructose-1-phosphate by ketohexokinase (KHK), which utilizes ATP [Articles:24065788, 23175570, 18398011]. This occurs primarily in the liver and results in a transient reduction of intracellular ATP and phosphate levels [Articles:24065788, 23175570]. Subsequently, adenosine monophosphate (AMP) deaminase is activated and AMP formed from the metabolism of fructose enters the purine catabolism pathway, ultimately resulting in uric acid as the end product [Articles:24065788, 23175570]. The hypothesis is that recent increases in sugar (and fructose) consumption in different countries correlates with an increase in gout, metabolic disease and diabetes and can in part be contributed to the result of higher uric acid levels [Articles:23175570, 19151107, 22198943, 23493538]. However, whether fructose intake causes clinically significant increases uric acid levels and subsequently results in the development of metabolic disease or gout is a controversial topic and whether patients should be advised to reduce fructose consumption to reduce uric acid levels remains a matter of debate - see other articles for more details: [Articles:23175570, 24065788, 23793849, 22645377, 22457397, 18163396, 21889564]. To our knowledge there are currently no drugs targeting fructose uptake or metabolism for the treatment of hyperuricemia.
Pharmacodynamics
There are three main pharmacological strategies for the treatment of gout or hyperuricemia: 1) reduce the generation of uric acid, 2) increase the removal of uric acid, 3) reduce the reabsorption of uric acid. This pathway discusses allopurinol and febuxostat that act via the first mechanism and urate oxidase that acts via the second. Their mechanism of action upon the uric acid pathway is depicted in the figure above and described below. Genetic variants associated with response to these drugs are detailed. The uricosurics class of drugs that work by the third mechanism in the kidney are discussed in the Uricosurics Pathway, Pharmacodynamics.
Allopurinol
Allopurinol is a drug indicated for the treatment of gout, prophylaxis of hyperuricemia in patients undergoing chemotherapy, and prevention of kidney stones recurrence (drug label). Allopurinol and its metabolite oxypurinol are analogues of hypoxanthine and xanthine, respectively, and work by binding to and inhibiting xanthine dehydrogenase (XDH), preventing the formation of uric acid (see figure above) [Articles:20562597, 12646938, 16597166, 22665944]. Hypoxanthine and xanthine are cleared in the urine or reutilized in the synthesis of nucleotides and nucleic acids [Article:11934348]. Allopurinol may also have beneficial pain relief effects, possibly through an increase in adenosine production from hypoxanthine (effects that are inhibited by caffeine) [Articles:19133997, 19133987]. However, a downside to allopurinol treatment is that it does not clear existing high levels of plasma uric acid, and as xanthine is less soluble than uric acid it may result in xanthine kidney stones or xanthine nephropathy [Articles:11934348, 12646938].
Allopurinol may alter response to drugs taken concomitantly. The prodrugs azathioprine and 6-mercaptopurine are converted into active metabolites by hypoxanthine phosphoribosyltransferase 1 (HPRT1) or inactivated by XDH along with other enzymes (see the Thiopurine Pathway, PK/PD) [Article:22132961]. Combining allopurinol with thiopurine therapy enhances HPRT1 activity and correlates with a significant increase in the amount of active thiopurine metabolite [Article:22132961]. Allopurinol decreases clearance of methotrexate which is associated with an increased risk of mucositis [Article:19834958].
Febuxostat
Febuxostat (Uloric®) is a non-purine inhibitor of XDH that can bind to and inhibit both oxidized or reduced forms of XDH, and does not seem to interact with the purine or pyrimidine catabolism pathways (see figure) [Articles:20562597, 19247302, 20401506]. It is indicated for the treatment of hyperuricemia in patients with gout, but not for asymptomatic hyperuricemia. It is an alternative therapy for patients where allopurinol has been contraindicated due to allergic responses [Articles:22582566, 20401506, 19247302] and has been shown to be more efficacious at 80mg than standard allopurinol treatment in several studies in Americans, with comparable adverse event rates [Articles:22316106, 22436129, 20370912, 23929928], though allopurinol efficacy rates differ from that in Europeans [Article:20401506]. Febuxostat is contradicted in patients treated with azathioprine or mercaptopurine due to drug-drug interactions.
Urate Oxidase (Uricase)
One strategy for enhancing uric acid excretion is to add exogenous urate oxidase enzyme not expressed in humans. Urate oxidase breaks down uric acid to 5-hydroxyisourate, which is then spontaneously degraded to allantoin without the aid of enzymes (see figure) [Articles:12646938, 16597166, 20562597]. Allantoin has 5-10 fold increased solubility compared to uric acid, and thus is more readily excreted via the kidneys [Articles:11934348, 16597166, 12646938]. Uricozyme® is urate oxidase extracted from Aspergillus flavus, however due to product-related impurities it is associated with acute hypersensitivity reactions [Articles:11934348, 20562597]. A recombinant form of urate oxidase, rasburicase (known commercially as Elitek®, Fasturtec®, Rasuritek®), was developed to reduce the occurrence of these reactions [Articles:11934348, 20562597]. Rasburicase has been shown to be a more effective treatment compared to allopurinol, rapidly reducing uric acid plasma concentrations and exposure [Articles:9369411, 11157020, 11342423, 11694947, 20562597]. Rasburicase however is unsuitable for the treatment of gout due to its short half-life, thus pegloticase (or PEG uricase, Krystexxa®) is recombinant urate oxidase conjugated to polyethylene glycol that is thought to result in reduced immunogenicity and an increased half-life compared to rasburicase [Articles:20562597, 22198943]. There are recent concerns raised regarding the development of antibodies against PEG that can be found in healthy blood donors (likely due to increasing exposure to such compounds), which may affect the efficacy of PEGylated pharmaceuticals [Article:22931049].
Pharmacogenomics
HLA genes and allopurinol-induced hypersensitivity reaction
A small proportion (0.1 to 0.4%) of patients receiving allopurinol develop life-threatening severe cutaneous adverse reactions (SCARs) that include drug hypersensitivity syndrome (DRESS), Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). The allopurinol hypersensitivity reaction is completely distinct from the drug's intended pharmacodynamic pathway: SJS and TEN are thought to occur via cytotoxic T cell-induced keratinocyte death by a drug-specific, HLA class I-restricted and granulysin-mediated pathway [Articles:17620823, 19029983]. NK cells may also be involved [Article:19029983].
The HLA-B*5801 allele is currently the most well-established genetic association with allopurinol-induced SCAR and has been demonstrated in numerous populations (Table 1). The human HLA-B gene is the most polymorphic in the genome, with over 1500 alleles that are made up of many variants [Article:17620823]. Historically, these alleles were identified by antigen binding assays but more recently sequencing and genotype techniques are utilized (see Table 1). Single Nucleotide Polymorphisms (SNPs) that tag the HLA-B*5801 allele differ in different populations [Article:16998491]. The mechanism behind HLA-B*5801-specific allopurinol-induced hypersensitivity is not fully understood, however several hypotheses proposed for HLA-dependent T cell stimulation by drugs could be applicable [Article:22017685]. Major Histocompatibility Complex (MHC) class I molecules (encoded by HLA-A, -B and -C) are expressed by most nucleated cells and are involved in presenting intracellular self or foreign peptides to CD8+ cytotoxic T cells [Articles:22017685, 17620823, 22943588, 23232549]. When presenting self-peptides (peptides), the interaction with specific T-cell receptors (TCRs) is weak. However when presenting a foreign/neo peptide, an antigen-specific CD8+ T cell response is elicited, resulting in T cell proliferation and effector function activation (such as cytotoxicity against the presenting cell) [Article:22017685]. Allopurinol or oxypurinol may bind self-protein/ peptide to create a haptenated product which undergoes antigen processing and is presented specifically by HLA-B*5801 to activate antigen-specific T cells (the hapten concept) [Article:22017685]. It has been proposed that at low concentrations the signal is insufficient to activate T cells, but at higher doses or due to renal failure (both potential risk factors for allopurinol-induced SCAR, Table 1), increased concentrations of drug results in activation of T cells [Article:22943588]. Allopurinol/oxypurinol may interact with the HLA-B*5801-MHC-peptide complex and TCR directly at the cell surface, rather than undergoing antigen processing (the pi concept) [Articles:22017685, 24591375]. During folding of HLA-B*5801 in the endoplasmic reticulum, allopurinol/oxypurinol may become incorporated into the peptide-binding groove, potentially changing the repertoire of self-peptides HLA-B*5801 is able to present, resulting in alloreactivity due to the presentation of novel peptides (the anchor site modification/occupation model) [Article:22017685]. A recent in vitro study suggests evidence for the pi concept, and that oxypurinol has higher affinity for the HLA-*5801 molecule compared to allopurinol in docking experiments [Article:24591375].
A meta-analysis calculated the odds ratios for allopurinol induced SJS/TEN in HLA-B*5801 carriers as 96.6 compared to allopurinol-tolerant matched controls, or 79 compared to population controls [Article:21906289]. The American College of Rheumatology (ACR) guidelines recommend screening only patients who are in high risk populations (Koreans with stage 3 chronic kidney disease, Han Chinese and Thai patients), and those found to be positive for HLA-B*5801 should be prescribed an alternative drug [Article:23024028]. The Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines also recommend not using allopurinol in patients who are known carriers of HLA-B*5801 [Article:23232549]. The FDA-approved drug label for allopurinol currently carries no information regarding HLA genotype, despite FDA scientists publishing a study assessing the clinical usefulness of allopurinol pharmacogenetics and reporting a strong and highly significant association between HLA-B*5801 and likelihood of allopurinol-associated SCAR [Article:22118056].
In some populations (Han Chinese, Thai, Korean) HLA-B*5801 explains 80-100% of the SCAR cases whereas in other populations (Japanese, European) HLA-B*5801 explains around 55% of the cases [Articles:15743917, 18192896, 19018717, 19696695, 21301380, 21545408, 21912425] suggesting that there are additional risk alleles, genetic variants and other factors that are also important for SCAR. In vitro experiments also suggest that allopurinol/oxypurinol-induced reactions are not strictly restricted to the HLA-B*5801 molecule [Article:24591375]. Additional alleles associated with allopurinol-induced SCAR include: rs9263726 A (which is in complete linkage with HLA-B*5801 in Japanese patients), rs2734583, rs3094011, rs2844665 C, rs3815087 A, rs3130931 C, rs3130501 G, rs3094188 A, rs9469003 C, HLA-A*33:03, HLA-C*03:02, HLA-C*08:01, HLA-Cw3, HLA-A33, HLA-DR13, HLA-D3, HLA-Cw*0302, HLA-A*3303, HLA-DRB1*0301, rs3117583, rs1150793, rs2855804, rs2268791, rs1594, rs2304224 (details of each study are available on the allopurinol PGx Research page [Articles:21912425, 21801394, 21545408, 21301380, 15743917].
Febuxostat was associated with hypersensitivity in two reported cases of patients who previously had adverse reactions to allopurinol [Articles:22383358, 21724706]- to our knowledge the genetic mechanisms behind this have not been described, as yet. No pharmacogenetic studies related to febuxostat are currently found.
+Table 1: Reported associations between HLA-B*58:01 and risk of allopurinol-induced SCARs.+
| Study Type and reference | Cases | Controls | OR (95% C.I.) | P value | Patient Population | Race/ ethnicity | Typing and genotyping method |
|---|---|---|---|---|---|---|---|
| Case/ control study [Article:23669020] | n=7 (SJS, SJS ocular type, erythema exudativum multiforme (EEM) minor). | n=25 (no ADRs). | 65.6 (2.9-1497) | 9.733x10^-4 | Hyperuricemia. | Japanese. | PCR-rSSO and PCR-SBT. |
| Case/ control study [Article:23600531] | n=25 (SJS, SJS/TEN, DRESS). | n=23 allopurinol- tolerant. | 39.11 (4.49-340.51) | 5.9x10^-4 | Asymptomatic hyperuricemia, gouty arthritis or nephropathy. | Portuguese. | PCR-rSSO and SSO-HR typing kit. |
| Observational study in family members [Article:23280169] | n=1 male who developed SJS. | n=1 the brother of the case, allopurinol-tolerant. | NA | NA | Gout and essential hypertension. | Self-described Han Chinese. | 4-digit, high resolution DNA sequencing. |
| Case/ control study [Article:22348415] | n=20 (SJS, DRESS, TEN, EMM). | n=30 allopurinol-tolerant, treated for at least 1 year (not matched to cases). | 123.5 (12.8-1195.1) (OR was higher when EMM case was excluded). | <1x10^-4 | NA | Han Chinese (Hong Kong). | Sequence-based typing. |
| Case-control study [Article:15743917] | n= 51 patients with allopurinol-induced SCAR (SJS, TEN or HSS). | n= 135 allopurinol-tolerant (at least 6 months of treatment). | 580.3 (34.4-9780.9) | 4.7 x10^-24 | Hyperuricemia. Chronic renal insufficiency was significantly higher in cases vs controls, and gouty arthritis was the converse. | Han Chinese residing in Taiwan. | Sequence-specific oligonucleotide reverse lineblot. |
| Comparison of allele frequencies in a population. [Article:18192896] | n=31 patients with allopurinol-induced SJC or TEN. | NA | 61 (32-118) for the allele frequency of cases compared to the allele frequency found in the general European population. | <10^-8 | Mostly hyperuricemia. | Mixed population, mostly European. | OLERSUP SSP HLA-B kit , sequencing in three cases. |
| Comparison of allele frequencies in a population. [Article:19018717] | n=10 patients with allopurinol-induced SJS, TEN (one also treated with carbamazepine, another also treated with phenytoin). | NA | 40.83 (10.53-158.9) allele frequency compared to the frequency in n=493 healthy Japanese subjects. | <0.0001 | NA | Japanese. | Sequencing. |
| Case-control [Article:19696695] | n= 27 patients with allopurinol-induced SJS or TEN (within 3 months of treatment). | n=54 allopurinol-tolerant patients (>6 months treatment) from the same hospitals. | 348.3 (19.2-6336.9) | 1.6x10^-13 | Hyperuricemia, some patients with gouty arthritis. | Self-identified Thai or Thai-Chinese. | PCR with sequence specific primers, and sequence-based typing. |
| Case/ control [Article:21301380] | n=25 patients with allopurinol-induced SCARs (20 with DIHS, 5 with SJS/ TEN). | n=57 allopurinol-tolerant patients. | 97.8 (18.3-521.5) (cases verses tolerant control). | 2.45 x10-11 | Cases = patients with gouty arthritis or hyperuricemia related to chronic renal failure. Controls = patients with chronic renal failure. | Korean. | Direct DNA sequencing analysis. |
| Comparison of allele frequency in healthy individuals. [Article:21545408] | n=7 allopurinol-induced SJS/ TEN patients. | n=115 healthy individuals. | 13.625 (2.774-69.448) | 0.248 after correction for multiple comparisons. | NA | Caucasian, Northern Italy. | 4-digit allele level within the antigen binding domain. PCR-SSP. |
| Case/ control [Article:21393610] | n=16 patients with an allopurinol hypersensitivity reaction (9 had SCARs, 7 had simple rashes). | n=432 allopurinol-tolerant patients (60 days). | 179.24 (10.19-3151.74) SCARs patients vs tolerant. | <0.001 | Patients with chronic renal insufficiency who took allopurinol. | Korean. | Serologic HLA typing by microlymphocytotoxicity method for HLA-B*58. |
| Case/ control [Article:22909208] | n=38 allopurinol- induced MPE, DRESS, SJS/ TEN (within first 2 months of exposure). | n=63 allopurinol- tolerant (treated for >3 months with no cutaneous manifestations). | 580.07 (32.18-10456.80) | 7.01 x10^-18 | Hyperuricemia and gouty arthritis. (A higher frequency of chronic renal insufficiency was seen in cases). | Cases = from the Southern region of China, control = all Han Chinese. | Direct DNA sequencing. |
| Case report [Article:19483528] | n=1, an 8-year old girl who developed TEN. | NA | NA. A test revealed she had HLA-B*5801. | NA | Developed asymptomatic hyperuricemia due to anti-TB treatment and was treated with allopurinol. | German, Kenyan parents. | Not described. |
| Case report [Article:22901319] | n=1, a 65-year old male who developed DRESS 1 month after allopurinol treatment initiation. | NA | NA. A test revealed he had a HLA-B*5801 positive genotype. | NA | Hyperuricemia. | Han Chinese. | Method not described. |
| Study identified patients across Australia with allopurinol hypersensitivity and carried out genotyping. [Article:21790926] | N=11 patients with allopurinol hypersensitivity including SJS/TEN and DRESS/DIHS, n=12 patients with MPE. | NA | NA. HLA-B*5801 was found in 6/5 cases with SJS/Ten, 1/5 cases with DRESS/DIHS and none of the patients with MPE. | NA | Not described. | Australian, mixed population: Caucasian and South-East Asian. | Four-digit high resolution DNA sequence-based HLA typing. |
| Case report [Article:17587850] | n=1, a 57-year old male who was diagnosed with SJS 10 days after allopurinol treatment. | NA | NA. Typing showed he had HLA-A31, A33, B51 and B58. | NA | Not described. | Not described. | Reverse sequence-specific oligonucleotide with PCR for serological HLA typing. |
| Case report [Article:17587850] | n=1, a 77 year-old male who was diagnosed with allopurinol-induced DIHS. | NA | NA. Typing showed he had HLA-A31, A33, B39 and B58. | NA | Not described. | Not described. | Not described. |
| Case report [Article:17587850] | n=1, a 93-year old male diagnosed with Allopurinol-induced SJS/TEN (a year earlier he had previously experienced maculopapular-type eruption and fever within 1 month of allopurinol treatment). | NA | NA. He had HLA-A24, A33, B52 and B58. | NA | Not described. | Not described. | Not described. |
| Drug-surveillance programme assessing Allopurinol cutaneous ADRs [Article:22017528] | n=84 cases, including maculo-papular eruptions, SJS, TEN and DRESS. Testing for HLA-B*5801 was only done in a subgroup of patients with SJS/TEN. | Allele frequency in a general European population of 0.015 was compared. | 18/18 cases of SJS/TEN in whom the assay was performed carried HLA-B*5801. OR=65.07 (30.66-138.09) compared to the general European population allele frequency | <0.0000. | Asymptomatic hyperuricemia, gout or secondary hyperuricemia (many also with hypertensive heart disease and renal failure). | Southern Sardinia, Italian, European. | PCR-SSO/ PCR-SSP. |
Urate oxidase and genes involved in oxidative stress regulation
Rasburicase and pegloticase are contraindicated in G6PD deficient individuals due to an increased risk of hemolytic anemia and methemoglobinemia, conditions involving red blood cells (RBCs) (link to drug label) [Article:15862084]. When rasburicase breaks down uric acid into allantoin, hydrogen peroxide (H2O2) is released as a by-product, a reactive oxygen species that can cause damage in RBCs and may ultimately result in cell turnover (hemolysis). Under normal conditions, the H2O2 is reduced to water and oxygen molecules by regulatory mechanisms, many of which require NADPH (See the Oxidative Stress Regulatory Pathway) [Articles:23913015, 16597166, 12646938, 15862084, 8704218, 2633878, 18177777, 16204390]. Methemoglobin (MetHb) is formed through the oxidation of heme iron atoms in hemoglobin and cannot transport oxygen or carbon dioxide; it is maintained at levels of around 1% by several controls (some of which require NADPH), in order to prevent methemoglobinemia (>1% MetHb) which can lead to cyanosis and tissue hypoxia (see the Methylene Blue Pathway, PD) [Articles:23913015, 23913015, 22024786, 7073040, 15862084, 10533013]. G6PD is an enzyme in the pentose phosphate pathway (PPP) that generates NADPH. This pathway is the only supply of NADPH in RBCs (see the Pentose Phosphate Pathway) [Articles:18177777, 2633878, 4154443, 15862084, 21376665, 16204390]. G6PD deficient RBCs are unable to enhance the PPP capacity to increase NADPH production and are thus more susceptible to oxidative damage that can ultimately result in methemoglobinemia and/or hemolysis [Articles:4154443, 2633878, 15862084, 22024786, 7073040, 8704218, 18177777, 16204390]. More than 180 genetic variants within the G6PD gene have been described to date, and many confer deficiency of the G6PD enzyme in RBCs [Articles:22293322, 17611006, 5316621, 2633878]. They are classified into 5 groups by the World Health Organization, depending on the degree of enzyme deficiency and associated clinical manifestations conferred; with Class I associated with Chronic Non-Spherocytic Hemolytic Anemia (CNSHA), Class II less than 10% of normal G6PD levels, and Class III 10-60% [Articles:5316621, 2633878] (see the G6PD VIP summary for more detailed information) [Article:22237549].
Several cases of methemoglobinemia and hemolytic anemia subsequent to treatment by rasburicase or urate oxidase have been reported in G6PD deficient individuals, though the underlying G6PD variant is often not reported and few studies report genotyping (Table 2). CPIC guidelines recommend avoiding the use of rasburicase in patients homo/hemizygous for G6PD variants that confer deficiency - in all other patients an enzyme test for G6PD deficiency is recommended prior to rasburicase use [Article:24787449].
Deficiencies in anti-oxidant mechanism pathways, not just in the G6PD enzyme, may further contribute to risk [Articles:17729111, 23913015]. Individuals with acatalasemia are homozygous for a catalase gene (CAT) variant that affects catalase mRNA expression (Japanese Type I; OMIM 115500.0001, II; OMIM 115500.0002, Hungarian Type A; OMIM 115500.0003, B-D) or have the unstable Swiss protein variant, resulting in minimal catalase activity in erythrocytes [Articles:1999334, 19122680]. Heterozygotes of Japanese and Hungarian CAT variants have hypocatalasemia, with around half of the normal catalase blood levels [Articles:19122680, 1999334]. Takahara's disease can develop in patients with acatalasemia from the production of H2O2 by oral microorganisms [Article:1999334]. Higher levels of methemoglobin are seen in Japanese acatalasemia erythrocytes compared to normal erythrocytes when exposed to nitrogen monoxide or dioxide [Article:19122680], and treatment of murine acatalasemia erythrocytes with H2O2 induces hemolysis in vitro [Article:16751193]. Methemoglobinemia developed in a patient with the CAT Japanese Type genetic variant when H2O2 disinfectant was used pre-surgery [Article:15220799]. In conclusion, catalase deficient patients may be more susceptible to methemoglobinemia and hemolysis with rasburicase treatment due to the release of H2O2, though direct cases have yet to be reported to our knowledge [Article:17729111]. Cases of individuals with combined G6PD and catalase deficiency have been reported [Article:1999334] and thus these individuals may be at a higher risk of rasburicase-induced methemoglobinemia and/or hemolysis.
Patients with cytochrome b5 reductase (CYB5R3, NADH-dependent methemoglobin reductase) deficiency are less able to reduce methemoglobin, and may be more susceptible to drug-induced methemoglobinemia and hemolysis [Articles:11418378, 31928, 6765904, 6620333, 5686480], along with individuals with hemoglobin variants e.g. Hasharon [Articles:6765904, 2069219], and deficiency in glutathione reductase (GSR) [Article:2069219].
+Table 2: Urate oxidase/rasburicase-induced adverse reactions associated with G6PD deficiency.+
| Patient details | Reason for urate oxidase/ rasburicase treatment | Consequence of rasburicase treatment | Genetic or enzyme screening | Reference |
|---|---|---|---|---|
| 16 year-old African American male. | Hyperuricemia due to Burkitt's lymphoma. | Developed methemoglobinemia. This was reversed by discontinuation of rasburicase, blood transfusion and oxygen delivery. | Diagnosed with G6PD deficiency because of known sensitivity - no screening to confirm was carried out and no genetic information was provided. | [Article:22190578] (Ng et al, 2011) |
| Adult case report. | Cancer patients at high risk of developing TLS. | Patients with known G6PD deficiency were excluded from the study, however one patient developed methemoglobinemia and hemolytic anemia after the first dose of rasburicase, which was discontinued. Methylene blue treatment exacerbated hemolysis, and the patient was identified as having G6PD deficiency. | Diagnosis method not reported and no genetic information provided. | [Article:22015451] (Vadhan-Raj et al, 2011) |
| Male adult case report | A male with Burkitt's lymphoma prescribed rasburicase before initiating chemotherapy. | Developed acute intravascular hemolysis. | The initial assay obtained soon after hemolysis was normal, however A- G6PD deficiency was later confirmed by a subsequent assay. | [Article:20196170] (Bain et al, 2010) |
| 12 year old Laotian male with T-cell ALL. | Hyperuricemia. | Developed methemoglobinemia and was treated with methylene blue. This did not improve methemoglobin levels or oxygen saturation levels and an exchange transfusion was undertaken to remove methemoglobin. | G6PD activity assay revealed a deficiency (genotyping not reported). | [Article:18561168] (Bhat et al, 2008) |
| African American male adult. | Acute renal failure secondary to hyperuricemia. | Developed methemoglobinemia and hemolytic anemia. Treated with daily packed red blood cell transfusions for 3 days, with resolution of methemoglobinemia and hemolysis. Rasburicase was thought to be the probable cause, as indicated by the Naranjo probability scale. | G6PD deficiency was confirmed by G6PD activity assay (no genotyping reported). | [Article:16204390] (Browning and Kruse, 2005) |
| Male adult. | Long term treatment with an i.v. of urate oxidase every other day for hyperuricemia several months following a kidney transplant. | 9 months later treated for 3 consecutive days with urate oxidase and subsequently diagnosed with hemolytic anemia, for which the patient received hemodialysis and recovered. | G6PD deficiency was later confirmed. The patient was classified as having the Mediterranean variant because of ethnic origin (born in Italy), although this was not confirmed by electrophoresis or genotyping. | [Article:2019023] (Ducros et al, 1991) |
| Pediatric case (mentioned in the discussion as an unpublished observed case). | Undergoing chemotherapy. | Hemolytic anemia. | Not detailed. | [Article:2019023] (Ducros et al, 1991) |
| A 12 year old African American male with ALL. | Patients with known G6PD deficiency were excluded from the study. | Developed methemoglobinemia after administration of non-recombinant urate oxidase (Uricozyme) and was later diagnosed with G6PD deficiency. | No test for diagnosis or genotyping reported. | [Article:9369411] (Pui et al, 1997) |
| A single case reported in the cohort of n=410. | A retrospective study of medical records of patients with B-cell lymphoma or ALL. | A single patient developed hemolysis with urate-oxidase treatment, 'revealing a previously unknown G6PD deficiency'. | No tests confirming the diagnosis of G6PD deficiency were mentioned. | [Article:12075750] (Patte et al, 2002) |
| African American adult male with multiple myeloma admitted for chemotherapy. | Hyperuricemia and acute kidney injury. | Methemoglobinemia. As G6PD status was unknown, ascorbic acid was administered to treat the methemoglobinemia rather than methylene blue. The patient developed hemolysis, and required red blood cell transfusion. | Test results on day 3 revealed the patient was G6PD deficient (by enzyme activity level, measured in U/g Hb). Both methemoglobinemia and hemolysis were attributed to G6PD deficiency (genotyping not reported). | [Article:22573495] (Sonbol et al, 2012) |
| A 14 year old Cambodian male diagnosed with Burkitt's lymphoma. | Received allopurinol for hyperuricemia, and furosemide and calcitonin for hypercalcemia, Rasburicase was administered before induction of chemotherapy. | Developed methemoglobinemia and subsequent hemolysis and required 3 red blood cell transfusions. | On day 3, G6PD deficiency was confirmed by quantitative assay (prior to the first transfusion). | [Article:17387701] (Borinstein et al, 2007) |
| A pediatric case in a cohort of n=278 pediatric and adult patients. | Rasburicase was administered in patients at risk of tumor lysis during initiation of chemotherapy in a compassionate use study. Patients with known G6PD deficiency were excluded. | A pediatric patient developed methemoglobinemia and was subsequently diagnosed with G6PD deficiency. | Testing method was not reported. | [Article:12942574] (Bosly et al, 2003) |
| A newborn male who was born at 30 weeks gestation by cesarean. | Among other complications, he was treated with rasburicase for a high uric acid level. | Hemolysis developed and he subsequently died. | A postmortem blood analysis showed a deficiency in G6PD enzyme, and genetic analysis revealed he was hemizygous for the G6PD Mediterranean variant. | [Article:23209099] (Zaramella et al, 2012) |
| 46-year old man, mixed Mauritian-Chinese background. | Treated for chronic lymphocytic leukemia. | 12 hours after rasburicase administration methemoglobinemia and hemolytic anemia developed. He was transfused with packed red blood cells, fluids and ascorbic acid. | Screening test revealed he had abnormal G6PD deficiency (variant not described). | [Article:23860572] (Cheah et al, 2013) |
| Case report of an adult male. | Part of a chemotherapy protocol. | A deficiency in G6PD was revealed. | Mediterranean variant determined by genotyping. | [Article:19654083] (article in French) (Joly et al, 2009) |
Summary
Unlike other animals, humans and some apes do not have the ability to break down uric acid into a more soluble form. High levels of plasma uric acid can be detrimental, contributing to several pathologies. Different pharmaceuticals have been developed to either inhibit the formation of uric acid, or increase its excretion. Here we have outlined the mechanisms of uric acid lowering drugs and detailed the genetic variants associated with adverse reactions to these drugs.
Edit history (3)
- 2012-10-04 Create
- 2014-06-12 Update Added publication
- 2024-08-28 Update fixed typos