About this pathway
Background
Tacrolimus (FK506) and cyclosporine (cyclosporin A, CsA) are cornerstone immunosuppressive agents given to solid organ transplant recipients to prevent and treat allograft rejection. The discovery of cyclosporine in the 1970s, and its entry into the collection of immunosuppressants in the early 1980s, was a major breakthrough in medicine. Cyclosporine was the most successful anti-rejection drug to date, and it radically improved the chance of survival for transplant recipients. In 1994, the Food and Drug Administration (FDA) approved tacrolimus, an effective alternative to cyclosporine [Article:15041303]. Since then, tacrolimus and cyclosporine have become the principal immunosuppressive drugs for solid organ transplantation. The United States Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients show that in 2011, 86% of the 16,055 patients who received a kidney transplant were prescribed tacrolimus upon discharge, and 2.4% were prescribed cyclosporine. One year after transplant, 84% and 4% of patients were receiving tacrolimus and cyclosporine therapy, respectively. Global differences exist in the usage of tacrolimus and cyclosporine: 2008 figures from the Australia & New Zealand Dialysis and Transplant Registry show that 61% of the 391 Australian patients who received a deceased kidney donor graft were prescribed tacrolimus, and 35% were prescribed cyclosporine. At one-year post transplant, these numbers changed to 55% and 33% for tacrolimus and cyclosporine, respectively. Tacrolimus and cyclosporine are also prescribed for liver, intestinal, lung and heart transplant recipients, and can be used to manage severe autoimmune conditions, such as atopic dermatitis [Articles:15175770, 14522634] and rheumatoid arthritis [Articles:15187241, 8448639].
Tacrolimus and cyclosporine differ in their chemical structure: cyclosporine is a cyclic endecapeptide [Article:8513650], while tacrolimus is a macrocyclic lactone [Article:8588225]. However, they act in a similar manner. Both are calcineurin inhibitors; their main mechanism of action involves inhibition of this important phosphatase [Article:15041303]. Tacrolimus exhibits similar effects to cyclosporine, but at concentrations 100 times lower [Article:2445722]. Despite these differences in potency, tacrolimus and cyclosporine both show excellent survival rates for grafts across many comparative studies (summarized in Maes et al. [Article:15041305]). However, several studies have shown that use of tacrolimus is associated with a lower allograft rejection rate compared to cyclosporine [Articles:16686762, 15741208, 16157605].
Pharmacokinetics
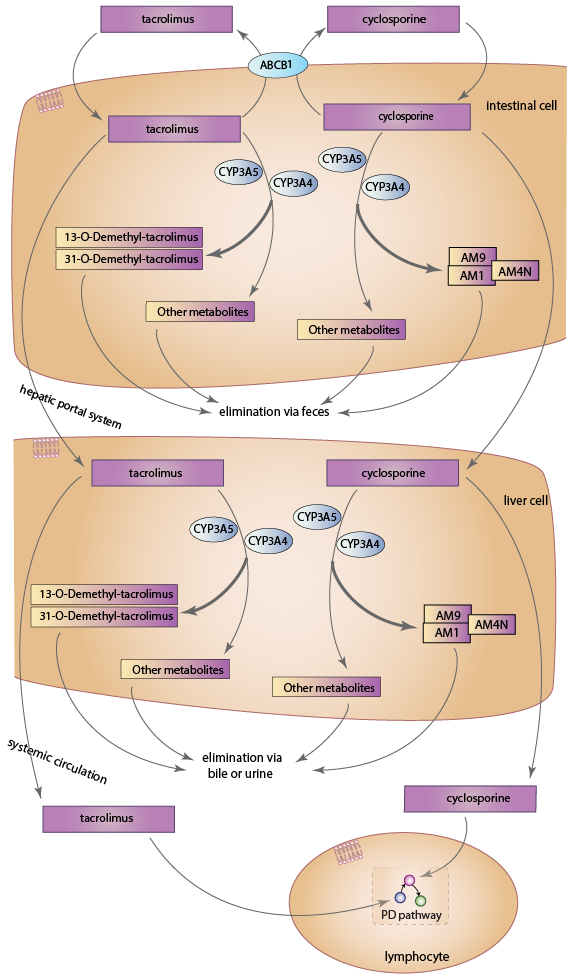
Upon entering enterocytes, both drugs are metabolized by gastrointestinal CYP3A isozymes, predominantly CYP3A4 and CYP3A5 [Articles:18978522, 22871995]. Studies have shown that CYP3A5 is the predominant enzyme for metabolism of tacrolimus, with CYP3A4 contributing, but having a lower efficiency for catalysis [Articles:16501005, 15951320]. In contrast, cyclosporine is primarily metabolized by CYP3A4 [Article:15450954]. The CYP3A family of enzymes also includes CYP3A7 and CYP3A43 [Article:12406645]. However, the involvement of CYP3A7 in cyclosporine metabolism is unclear [Article:18978522], and it has a low affinity and capacity toward tacrolimus, suggesting that it likely plays a minimal role in tacrolimus metabolism [Article:15951320]. Similarly, there is no evidence to support the involvement of CYP3A43 in cyclosporine metabolism [Article:18978522], and its role in tacrolimus metabolism, if there is one, has yet to be elucidated [Article:15244495]. Parent drug that escapes intestinal metabolism enters the hepatic portal system and the liver, where CYP3A4 and CYP3A5 metabolize tacrolimus and cyclosporine [Articles:8689938, 1683920]. Upon entering systemic circulation, both drugs bind extensively to erythrocytes [Articles:15041303, 6129301], and only unbound drug is capable of entering lymphocytes and exerting its main immunosuppressive effects.
Up to fifteen metabolites of tacrolimus may be formed [Article:17965516], with the most prevalent being 13-O-Demethyl-tacrolimus [Articles:7507815, 15244495]. This metabolite is approximately one-tenth as active as tacrolimus, while a minor metabolite, 31-O-Demethyl-tacrolimus, has been found to have an immunosuppressive activity comparable to tacrolimus [Articles:7507815, 17965516]. The remaining metabolites have all been found to have weak or negligible pharmacological activity [Articles:7536652, 17965516]. For cyclosporine, approximately 25 metabolites are formed [Article:22210099]. The major metabolites found in blood are AM1 and AM9, which are hydroxylated products, and AM4N, which is N-demethylated [Article:22210099]. CYP3A4 is capable of transforming cyclosporine into AM1, AM9 and AM4N, while CYP3A5 only transforms the drug into AM9 [Article:15450954]. Reported immunosuppressive activity of these metabolites varies between studies, but all metabolites studied so far have reduced activity compared to cyclosporine [Articles:2137384, 1521927]. AM1 has the highest immunosuppressive activity, with some studies finding its activity at close to 20% of native cyclosporine [Article:1521927], and others finding it as high as 80% [Article:2137384].
Both tacrolimus and cyclosporine are extensively metabolized, with less than 0.5% and 1%, respectively, of the parent drug appearing unchanged in the in urine and feces [Articles:10348790, 21151751]. Approximately 95% of tacrolimus metabolites are eliminated via the biliary route with urinary excretion accounting for around 2% [Article:10348790]. Similarly, cyclosporine metabolites are mainly excreted in the bile, with only around 3% of the drug undergoing renal elimination [Articles:21151751, 2587622].
In addition to CYP3A4 and CYP3A5, the efflux transporter P-glycoprotein also plays a major role in the pharmacokinetics of tacrolimus and cyclosporine [Article:10837558]. Encoded by the ABCB1 gene, it pumps xenobiotics from the cytoplasm to the exterior of the cell [Article:9333100]. It is present on the apical surface of cells, and is known to transport both tacrolimus and cyclosporine [Article:7681059]. P-glycoprotein has been found to be present at high concentrations in villus tip enterocytes of the small intestine [Article:10518631], and lowers intracellular concentrations of both drugs by pumping them out of enterocytes into the intestinal lumen [Article:9333100]. In addition to enterocytes, ABCB1 also transports drugs across membranes within hepatocytes [Article:16377077] and kidney cells [Articles:18518969, 9375821]. It is also involved in drug transport within lymphocytes [Articles:10716499, 17606483], so the actual concentration of cyclosporine and tacrolimus available within these cells may be influenced by the cellular P-glycoprotein content. Nevertheless, P-glycoprotein's role within enterocytes is better characterized, and so only its involvement in intestinal drug transport is shown. Variation in intestinal P-glycoprotein accounts for approximately 17% of the variability in oral clearance of cyclosporine, where higher levels of P-glycoprotein indicated higher observed clearance of the drug. Indeed, the same study concluded that 75% of inter-patient variability in cyclosporine clearance could be explained by variation of both CYP3A4 activity in the liver, and expression of P-glycoprotein in enterocytes [Article:9333100]. For tacrolimus, a strong inverse correlation was seen between the concentration/dose ratio of tacrolimus and the intestinal mRNA level of ABCB1 for the first 7 days following liver transplant in one study [Article:15919446], and for the first 4 days following liver transplant in another [Article:16413244].
A number of drugs have been reported to interact with tacrolimus and cyclosporine. Comprehensive lists can be found in reviews by van Gelder [Article:12167066], Christians et al. [Article:12190331], and Campana et al. [Article:8906896]. Drug interactions mainly occur when tacrolimus or cyclosporine is co-administered with either inhibitors or inducers of cytochrome P450 3A (CYP3A) or P-glycoprotein [Article:12190331], two proteins that have significant overlap in substrate specificities [Article:7619215].
Pharmacogenomics
The majority of pharmacogenetic studies on tacrolimus and cyclosporine have focused on the effects of variants in the CYP3A4, CYP3A5 and ABCB1 genes because of the central role the enzymes and transporters they code for play in tacrolimus and cyclosporine disposition. However, a few studies have examined the influence of single nucleotide polymorphisms (SNPs) within the gene encoding the pregnane X receptor (NR1I2), which regulates the expression of multiple genes including CYP3A and ABCB1 [Article:23095803]. Additionally, a couple of studies have examined SNPs in the POR gene, which encodes for CYP450 oxidoreductase, a protein responsible for transferring electrons from NADPH to CYP450 enzymes, enabling their activity [Article:11371558]. Several studies have also looked at variations in the TGF-ß1 gene (TGFB1), the cyclophilin A gene (PPIA), and the CYP2C8 gene; CYP2C8 is involved in the metabolism of arachidonic acids (AAs) into epoxyeicosatrienoic acids (EETs), metabolites implicated in maintaining normal renal function. Despite these numerous studies, only the *3 allele (rs776746) in the CYP3A5 gene has shown strong associations with tacrolimus pharmacokinetics. Very little consistent evidence has emerged for factors affecting tacrolimus pharmacodynamics or cyclosporine pharmacokinetics and pharmacodynamics. The overall inconsistency of these studies may be related to ethnic variability, small numbers of patients, non-specific pharmacokinetic assays, variation in when outcomes are measured, and the impact of donor genotype - particularly in nephrotoxicity studies in kidney transplant patients or pharmacokinetic studies in liver transplant patients. Larger studies and meta-analyses that take ethnicity and donor genotype into account may help resolve some of this variability.