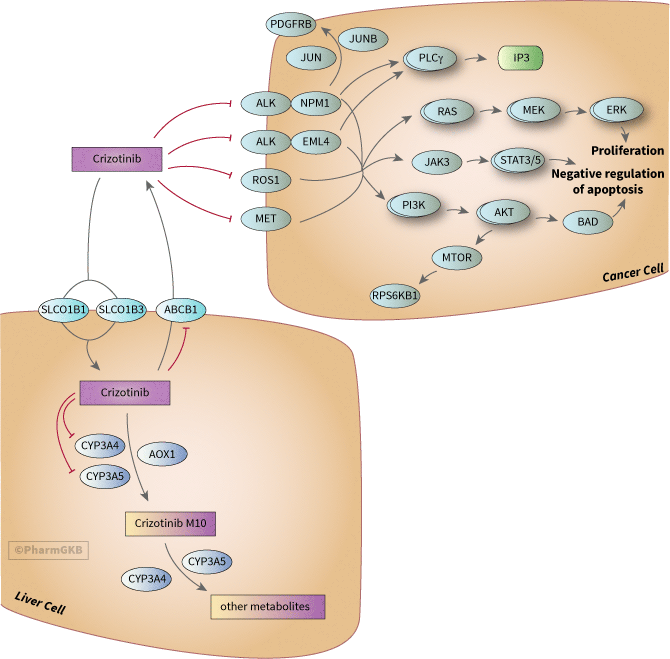
About this pathway
Background
Crizotinib is an orally active tyrosine kinase inhibitor approved by US-FDA for treatment of patients with locally or advanced non-small cell lung cancer (NSCLC) with ALK alteration or ROS-1 alteration. It is a selective inhibitor of ALK (anaplastic lymphoma receptor tyrosine kinase) and mesenchymal epidermal growth factor (MET) [Article:20979469]. Crizotinib is also effective against ROS1 rearranged NSCLC; ROS1 is 77% homologous to ALK in kinase domain [Articles:25264305, 27328934] NSCLC response rates for crizotinib are greater than 60% with a mean progression-free survival of over 7-10 months prior to acquisition of resistance to crizotinib monotherapy [Article:24419423]. Of all patients with NSCLC, around 4% have ALK variants, and about 1-2% have variants of ROS1 or MET [Article:23401436]. Patients with ALK variant NSCLC tend to be non-smokers, younger than average and the tumors are adenocarcinomas [Article:23401436].
Pharmacodynamics
The pharmacodynamics of crizotinib are based on the signaling pathways of the tyrosine kinases that it binds. These signaling pathways have a great deal of overlap downstream and result in cell cycle dysregulation and proliferation.
The ALK gene encodes a transmembrane receptor tyrosine kinase belonging to the insulin receptor superfamily. ALK expression in humans is normally limited to cells of neural origin [Article:17519389]. The ALK gene was originally identified in Anaplastic Large Cell Lymphoma ALCL but is now known to be involved in over sixteen types of cancers including NSCLC and neuroblastoma [Article:24091028]. The oncogenic potential of ALK is triggered by chromosomal rearrangement resulting in the formation of fusion products with several genes like EML4 and NPM1 or multiple copies of ALK [Articles:18166835, 21423761, 24750504]. EML4-ALK fusions are most common ALK fusion in NSCLC [Article:24091028]. There are at least nine different variants of EML4-ALK fusion gene, which vary in their break points [Article:23401436]. EML4-ALK fusion proteins mediate ligand-independent oligomerization of ALK resulting in constitutive ALK kinase activation, which subsequently results in uncontrolled cell proliferation via activation of phosphoinositide-3 kinase (PI3K), mitogen activated kinase (MAPK) and jun kinase (JAK) pathways [Article:21288922]. Some of the main gene groups involved in downstream signaling of ALK-EML4 are shown in the figure; for a more detailed signaling pathway of NSCLC see KEGG.
ALK-NPM1 fusions are the most common ALK alteration in ALCL [Article:23064464]. ALK-NPM1 fusions lead to up-regulated JUN, JUNB and PDGFRB, which can sensitize tumors to imatinib even those tumors with ALK that has mutated and become resistant to crizotinib [Article:23064464]
As with ALK, ROS1 variants are also chromosomal rearrangements and fusions of the gene to other genes; the most common gene partners observed in NSCLC being CD74 (42%), EZR (15%) and SLC34A2 (12%)[Article:23814043]. Pathways downstream of ROS1 that become activated are the PI3K/AKT/mTOR, JAK/STAT, and MAPK/ERK pathways that promote proliferation and cell survival [Article:23814043].
ALK and ROS rearrangements are not present in same tumors (reviewed in [Article:23814043]). Also ALK-EML4 rearrangements are in mutual exclusion with EGFR and KRAS mutations [Article:21904575]. ROS1 rearrangements are associated with younger age, never-smoking history, Asian ethnicity, and advanced stage and not generally found with other EGFR and KRAS mutations. However, one study of 556 NSCLC tumor samples found one ALK positive and two ROS1 positive that also had mutations in EGFR [Article:22661537].
Activation of MET also increases signaling via PI3K/AKT, RAS/RAF/MAPK and STAT [Article:23423860]. In NSCLC, amplification of MET gene was associated with shorter survival after surgery [Article:19255323]. Amplification of MET has been observed as a mechanism of resistance to EGFR pathway treatments such as gefitinib, making crizotinib a possible second line of therapy in these cases [Article:28138027].
Acquired Resistance
The initial response rate for NSCLC patients to crizotinib is approximately 60% and had improved PFS compared to chemotherapy standard second-line therapies such as pemetrexed and docetaxel; however many patients develop acquired resistance within the first year (reviewed in [Article:28054318]). Resistance to crizotinib can occur in a variety of ways: secondary mutations in ALK, copy number increases in ALK, activation of other pathways that bypass ALK inhibition, and progression of the disease in the central nervous system (CNS) where the drug does not penetrate well [Article:28054318].
The first crizotinib resistance mutations reported in ALK were tyrosine kinase domain at C1156Y and in the “gatekeeper position” at L1196M [Article:20979473]. The “gatekeeper” is a term used in resistance to other TKIs that is at the bottom of the ATP binding pocket and corresponds to EGFR position 790 and position 315 in ABL [Article:20979473]. Additional mutations include L1152R, I1151Tins, F1174C/L/V, G1202R, G1202del, D1203N, and S1206Y/C and are reviewed in [Article:28122866]. ALK mutations represent around 20-30% of crizotinib resistance [Article:28122866]. Second generation ALK inhibitors such as ceritinib, alectinib and brigatinib have differential sensitivities to crizotinib-resistant variants of ALK [Article:28077299]. Ceritinib overcomes some crizotinib-resistant variants in ALK I1171T, L1196M, S1206Y, and G1269A mutations, but not F1174C or G1202R mutations; alectinib is effective against L1196M, C1156Y and F1174L ; brigatinib is effective against C1156Y, F1174L, L1196M, G1202R, and R1275Q [Article:28077299].
Amplification of ALK occurs in between 6-18% of crizotinib resistant cases [Article:28122866].
EGFR activation was the first example of crizotinib-resistance developed via activation of bypass pathways [Article:21791641]. Other EGFR family members such as ERBB3 and ERBB4 have also been identified in resistance via parallel pathway activation [Article:28122866].
Pharmacokinetics
Crizotinib M10, crizotinib lactam is the major metabolite of crizotinib in humans [Article:25034009]. No Pubchem record was found for this metabolite but the structure is depicted in Figure 8 on page 26 of the FDA document and in [Article:25034009]. It is a racemic mixture of two steroisomers, PF-06270079 and PF-06270080 [Article:25034009]. It is likely formed by a two step process of metabolism of crizotinib involving formation of an imine intermediate by enzymes CYP3A4, CYP3A5 and AOX1 [Article:26381275]. CYP3A4 and CYP3A5 are also involved in the metabolism of M10 to other metabolites [Article:26381275]. Structures of the other metabolites, which are minimally found in plasma, urine and feces, are also described in [Article:25034009] but not in PubChem. Crizotinib M10 had some activity against crizotinib targets in vitro but was 2.5- to 7.7-fold less potent than crizotinib against ALK and 2.5- to 4-fold less potent against MET [Article:25034009]. No other metabolites showed any activity against ALK or MET [Article:25034009].
Crizotinib exhibits time-dependent pharmacokinetics due to inhibition of CYP3A4; after repeat dosing there is a 40% decrease in oral clearance [Article:23129213]. The metabolites are primarily excreted in feces (63%) and 22% in urine [Article:25034009]. Co-administration of crizotinib with strong CYP3A inducers may decrease crizotinib exposure. In healthy volunteers, co-administration of single dose of crizotinib with multiple doses of rifampicin was associated with increased clearance and decreased exposure [Article:26381275]. Conversely, co-administration of crizotinib with CYP3A inhibitors may increase crizotinib exposure. In healthy volunteers, co-administration of single dose of crizotinib with multiple doses of ketoconazole was associated with decreased clearance and increased exposure [Article:26381275]. Plasma concentrations of crizotinib M10 were also higher following co-administration of crizotinib with ketoconazole.
In vitro studies and mouse models indicate that crizotinib is a substrate of transporter ABCB1 but not ABCG2 [Article:24037730]. Crizotinib is also an inhibitor of ABCB1, impeding in vitro ABCB1-mediated transport of the substrate doxorubicin [Article:22233293]. As mentioned above, crizotinib does not cross the blood-brain barrier (BBB) well [Article:21422405]. ABCB1 is expressed at the BBB and known to limit the availability of other TKIs in the brain [Article:26579371], and perhaps may explain why crizotinib penetrates the CNS poorly. Studies in transfected HEK cells showed SLCO1B1 and SLCO1B3 are capable uptake of crizotinib [Article:23340295].
The most common treatment-related adverse events are reversible mild to moderate (grade 1 or 2) visual effects (including visual impairment, photopsia, blurred vision, photophobia, diplopia), nausea, vomiting, diarrhea, constipation, elevated ALT levels and neutropenia (grade 3-4) [Article:22191798].
Pharmacogenomics
To date there is only one paper that looks at pharmacokinetic PGx of crizotinib. A case series of 8 Asian patients with ALK positive NSCLC showed that AA homozygosity at any of the three locations of ABCB1*2 (rs1128503, rs2032582 and rs1045642) was associated with increased exposure to crizotinib [Article:27270784]. Increased exposure was significantly associated with toxicity [Article:27270784].
Reactions & interactions (36)
-
Activation
ALK-NPM1 → PLCgamma
-
Activation
ALK-NPM1 → AP-1
-
Activation
AP-1 → PDGFRB
-
Activation
PLCgamma → inositol 1,4,5-trisphosphate
-
Activation
ALK-EML4 → PI3K
-
Activation
MTOR → RPS6KB1
-
Activation
PI3K → AKT
-
Activation
ALK-EML4 → JAK3
-
Activation
MET → Ras
-
Activation
AKT → BAD
-
Activation
ALK-NPM1 → PI3K
-
Activation
ALK-EML4 → Ras
-
Activation
ALK-EML4 → PLCgamma
-
Activation
ALK-NPM1 → JAK3
-
Activation
ROS1 → Ras
-
Activation
JAK3 → Stat3/5
-
Activation
ALK-NPM1 → Stat3/5
-
Activation
AKT → MTOR
-
Activation
MEK → ERK
-
Activation
Raf → MEK
-
Activation
Ras → Raf
-
Biochemical Reaction
crizotinib → crizotinib M10
-
Catalysis
ABCB1 → Transport
-
Catalysis
SLCO1B1 → Transport
-
Catalysis
SLCO1B3 → Transport
-
Catalysis
CYP3A5 → Biochemical Reaction
-
Catalysis
AOX1 → Biochemical Reaction
-
Catalysis
CYP3A4 → Biochemical Reaction
-
Inhibition
crizotinib → MET
-
Inhibition
crizotinib → ABCB1
-
Inhibition
crizotinib → ROS1
-
Inhibition
crizotinib → ALK-NPM1
-
Inhibition
crizotinib → ALK-EML4
-
Inhibition
crizotinib → CYP3A4
-
Transport
crizotinib → crizotinib
-
Transport
crizotinib → crizotinib
Edit history (2)
- 2018-03-22 Create
- 2019-02-25 Update Update to new illustrator formatting.