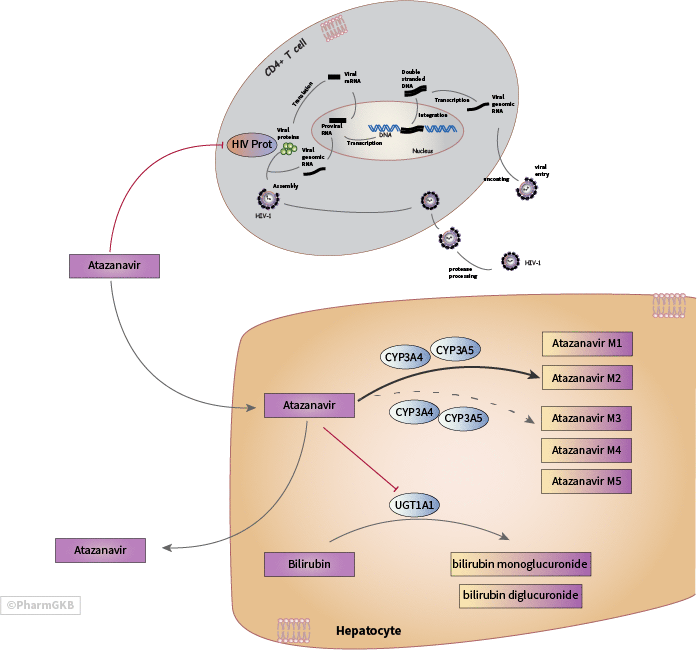
About this pathway
Introduction
Atazanavir (ATV) is an azapeptide of the protease inhibitor (PI) drug class because it selectively inhibits HIV genotype I (HIV-I) protease, an enzyme critical for HIV-1 virion maturation. ATV is typically administered in combination with a pharmacokinetic enhancer (also known as a “booster”) to improve its pharmacokinetic (PK) profile and two different nucleoside reverse transcriptase inhibitors (NRTIs) as part of combination anti-retroviral therapy (cART) [Articles:25151562, 25151564]. Once-daily-dosing, fewer metabolic side effects as compared to other PIs, and a high-barrier to drug resistance make ATV, particularly when boosted, a good option for HIV-1 positive individuals with poor adherence, cardiovascular diseases or diabetes mellitus [Articles:15585441, 18722869, 26372383] Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV. Genetic variants in the genes encoding the principal enzymes involved in the metabolism of ATV, which are also central to the metabolism of many commonly prescribed drugs, have been found to be associated with adverse events and changes in the pharmacokinetics of ATV. Common adverse effects associated with ATV include hyperbilirubinemia, cosmetic jaundice, nephrolithiasis and cholelithiasis [Article:26360703]. The updated Health and Human Services 2017 “Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents” now favors integrase strand transfer inhibitor (INSTI) regimens over PIs, including ATV, as the preferred initial regimen for treatment naïve patients and now considers ATV/ritonavir (ATV/r) with two NRTIs as a “Recommended, Alternative, and Other Antiretroviral Regimen Options for Treatment-Naïve Patients” but recognizes that it may still be “the preferred regimen for some patients”Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV.
Pharmacokinetics
ATV is rapidly absorbed and peak ATV concentration (Cmax) is reached between 2 – 2.5 hours post-dose. ATV absorption is improved when it is taken with food. The primary route of ATV elimination is through bile (79% of administered dose) and the secondary route is through urine (13% of administered dose excreted as metabolites). There are well-established pharmacokinetic differences when comparing between races, men and women, and HIV-1 positive subjects compared to HIV-1 negative subjects that have yet to be explained [Articles:15585441, 16176117]. In addition, there are also documented differences between men and women with respect to time to virologic failure, although study results are contradictory [Articles:25159623, 24253247, 25979186].
ATV is primarily and extensively metabolized by the cytochrome P450, family 3, subfamily A (CYP3A) polypeptide isoenzymes 4 and 5 (CYP3A4/5) [Article:16176117]. What is unknown is whether it enters cells through diffusion, or whether influx is mediated by specific transporters. The organic anion transporter protein 1B1 (OATP1B1), which is encoded by the solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene, is known to facilitate the uptake of conjugated-bilirubin from blood and possibly unconjugated bilirubin and ritonavir. According to the results of at least one in vitro study, ATV may be an inhibitor of OATP1B1 [Article:23750830]. However, the effect of any polymorphisms of SLCO1B1 in patients administered ATV may actually be due to the fact that ritonavir may be an inhibitor of OATP1B1, rather than ATV, although this is not yet established [Articles:20051929, 22541068, 20102298, 23750830]. ATV may be a substrate of P-glycoprotein (P-gp), which is encoded by the ATP-binding cassette sub-family B member 1 (ABCB1) gene, as well as an inhibitor although more evidence may be needed to clarify this relationship [Articles:20551216, 16355344, 22394315, 24997317, 23224979]. None of the studies discussed here have demonstrated a genetic association between ATV and the nuclear receptor subfamily 1 group I member 2 (NR1I2) gene that encodes the pregnane X receptor, variants in this gene have been investigated with respect to ATV PK, possibly because it regulates the expression of CYP3A4, SLCO1B1, and ABCB1 [Articles:18831695, 24997317, 20921307].
The US Food and Drug Administration (FDA)-approved drug label for ATV indicates that the major biotransformation pathways of ATV are mono- and di-oxygenation while minor biotransformation pathways include glucuronidation, N-dealkylation, hydrolysis, oxygenation and dehydrogenation Atazanavir Drug Label. Four studies investigating biotransformation pathways of ATV confirm that mono-oxidation is a major pathway of ATV biotransformation with two explicitly identifying the structures of one or two mono-oxidation products (M1, M2), such as “a hydroxylated phenylbutane moiety”, while two others solely identified multiple “mono-oxidated” metabolites of ATV without specifying their structures. All four studies detected the presence of metabolites that were the product of carbamate hydrolysis on the prime and non-prime sides of ATV (M3, M4), and three studies detected one N-dealkylation metabolite (M5) (Figure 1) [Articles:19546238, 21148252, 21148251, 23722954]. None of the studies, which assessed metabolites in human plasma, human liver microsomes, hepatocytes, or in mouse feces and urine, reported identifying glucuronidated ATV metabolites. In addition, no ATV glucuronide structures are deposited in PubChem, which contradicts information on the FDA-approved drug label. When unboosted, mono-oxidation and oral clearance (CL/F) of ATV may occur more rapidly in those individuals who carry at least one functional version of CYP3A5 as compared to individuals who carry no functional copies due to homozygosity of CYP3A5*3 (c.219-237 C>T at rs776746), CYP3A5*6 (c. 624 C>T at rs10264272), or CYP3A5*7 (c.1035_1036 insT at rs41303343) [Articles:26892777, 21148251, 19710077].
Ritonavir as a pharmacokinetic enhancer of Atazanavir Ritonavir is also a protease inhibitor, but its ability to block CYP3A-mediated metabolism of ATV makes it an effective pharmacokinetic enhancer (or “booster”). When boosted with ritonavir (300 mg ATV and 100 mg ritonavir; ATV/r), ATV’s pharmacokinetic profile is improved and individual differences in ATV pharmacokinetics are diminished [Article:16176117]. Treatment with unboosted ATV is approved by the FDA for specific cases although the European Medicines Agency (EMA) does not formally approve it [Article:22646049]. ATV administered without a pharmacokinetic enhancer is not a recommended course of therapy for treatment naïve individuals, individuals with a very high viral load (e.g. 100,000 > or = copies HIV-1 RNA /mL) Atazanavir Drug Label Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV, patients with previous virologic failure ( > 50 copies HIV-1 RNA/mL), or pregnant women. ATV without a pharmacokinetic enhancer may be recommended in special cases such as to reduce pill burden, improve treatment adherence, or to prevent long-term toxicities associated with ritonavir in individuals with hepatic impairment [Article:22646049].
The ATV drug labels advises against co-administration of unboosted ATV with tenofovir since tenofovir had been shown to decrease ATV exposure, mostly through increased clearance. However, current consensus regarding the effect of tenofovir on ATV exposure in subjects administered ATV/r is less clear: some studies continue to find that tenofovir is associated with a decrease in ATV exposure [Articles:22646049, 18025112, 21649692, 21283017, 21670182], while others report no significant negative effects on ATV exposure in patients administered ATV/r [Articles:24992294, 23392467, 19858926, 17665183, 19897506]. In a randomized, controlled pilot study in which patients were switched to ATV without ritonavir, a pharmacogenetically-informed dosing algorithm helped patients maintain ATV at therapeutic levels, despite co-administration with tenofovir [Article:26174719]. Interestingly, another study in Taiwanese patients undergoing therapeutic drug monitoring (TDM) that had been switched to ATV without ritonavir from a ATV/r regimen actually found that 16.5% of the tenofovir group (N =103) but 35.1% patients without tenofovir (N = 94) had ATV concentrations below the recommended therapeutic values (P < 0.01). Time to virologic failure was not different between groups [Article:26857335]. It is important to note that in both studies, patients were only included if they had no history of virologic failure or resistance associated mutations (RAMs). A recent meta-analysis of five randomized controlled trials compared ART regimens including ATV versus ATV/r in treatment-experienced subjects for whom ritonavir was no longer advised. This study reported improved safety outcomes in the groups without ritonavir without sacrificing efficacy but the authors cautioned that longer-term studies of efficacy and safety were needed [Article:24314017].
Pharmacodynamics
HIV-1 protease The target of ATV is HIV-1 protease. ATV interferes with maturation of the HIV virion by blocking the active site of the HIV-1 protease so that it cannot cleave the viral gag and gag-pol polyproteins (Figure 1) [Article:23886005]. There is a high barrier to resistance with PIs, but certain resistance associated mutations (RAMs) in HIV-1 protease do confer resistance to ATV and increase susceptibility to the remaining PIs, notably isoleucine to leucine at amino acid residue 50 (I50L), the isoleucine to valine mutation at residue 84 (I84V), and the arginine to serine mutation at residue 88 (N88S). Knowing whether a patient carries one of these mutations is very helpful for clinicians to decide the best treatment course for a patient [Articles:15585441, 16921473, 28208121].
UGT1A1 Glucuronidation of substrates increases their water solubility and facilitates their excretion in bile and urine. The UDP glucuronosyltransferase (UGT) family of enzymes is responsible for the glucuronidation of a wide variety of endogenous and xenobiotic substrates. Among the 19 functional enzymes of the human UGT super family, only UGT family 1 member A1 (UGT1A1) is known to be capable of glucuronidating bilirubin, a product of heme catabolism. Since ATV is a competitive inhibitor of UGT1A1, a common adverse effect in patients administered ATV is elevation of non-glucuronidated (unconjugated) bilirubin, a condition known as hyperbilirubinemia. Since UGT1A1 does not metabolize ATV, this relationship appears to be an off-target PD interaction. Hyperbilirubinemia due to ATV is not necessarily indicative of hepatic injury but may result in cosmetic jaundice and there is concern that it may compromise drug adherence in certain patients [Articles:24492252, 26595536].
Drug-Drug Interactions
Many commonly prescribed drugs are metabolized, inhibited, or induced by the main ATV metabolizing enzymes CYP3A4/5 so there is a high potential for drug-drug interactions, especially in older patients where polypharmacy is common, and in patients co-infected with hepatitis C, tuberculosis, or other opportunistic bacterial or fungal infections, a common complication in individuals with HIV [Article:26604826]. A case series in four HIV positive patients reported that ATV and ritonavir Cmin and AUC were only moderately reduced in patients administered voriconazole, a triazole antifungal agent [Article:25155930]. A recent study in healthy volunteers reported that co-administration of ATV/r with voriconazole also lead to reductions of ATV Cmin of between 20-30% regardless of the metabolizer phenotype of the primary metabolizing enzyme of voriconazole, CYP2C19. Although CYP2C19 is the primary metabolizing enzyme, voriconazole is also metabolized by CYP3A4 [Article:28277330]. The study also found that co-administration of voriconazole and ATV/r led to decreased voriconazole exposure in subjects with the CYP2C19 extensive metabolizer phenotype, and increased voriconazole exposure in the CYP2C19 poor metabolizer phenotype, which the authors attribute to CYP2C19 induction by ritonavir [Article:27432796]. The FDA-approved drug label for ATV also gives specific dose scheduling and dosing recommendations for patients who are concomitantly taking H2 receptor antagonists, proton-pump inhibitors (PPIs), antacids, or buffered didanosine because ATV absorption depends on low gastric pH [Articles:15585441, 16176117, 24595498]. Other drugs that are extensively metabolized by UGT1A1, such as the cancer drug irinotecan, are contraindicated due to the high potential for toxicity Atazanavir Drug Label [Article:15585441].
Pharmacogenomics of ATV/r
UGT1A1 Gilbert’s Syndrome is characterized by mild and intermittent elevations of bilirubin caused by homozygosity of the c.-53_-52 (TA)6 > (TA)7 allele in UGT1A1 at rs8175347 (*28). The UGT1A1 *28 allele consists of seven thymidine- adenine (TA) tandem repeats in the promoter region of UGT1A1 where normally there are six (UGT1A1 *1 allele). The *28 allele causes a ~50% decrease in UGT1A1 protein expression. Similarly, the *37 ((TA) 8) allele also decreases UGT1A1 transcriptional activity relative to *28, whereas the *36 ((TA)5) allele in UGT1A1 leads to increased transcriptional activity relative to *28 [Article:26595536]. The *36 and *37 alleles are rare in White and Asian populations, but are more common in West and sub-Saharan African populations [Article:21309756]. The UGT1A1*6 allele (c.211 G>A at rs4148323), which causes a missense mutation (G71R), is more prevalent in individuals of East Asian descent but has not been found to be associated with ATV-associated hyperbilirubinemia [Article:20504240].
The UGT1A1 *28/ *28 genotype has been repeatedly shown to be associated with hyperbilirubinemia in patients administered ATV and there is some evidence that the homozygous genotype may also be associated with drug discontinuation [Articles:17058217, 19710077, 23148286, 16170755, 21288825, 17148966, 20504240, 26417955]. Most recently, a study in 321 patients reported an association between the T allele at rs887829 in the promoter region of UGT1A1 (c.-364 C>T) and bilirubin-associated discontinuation. The hazard ratio (HR) for bilirubin-related discontinuation for all subjects with the TT genotype (vs. CC genotype) was 7.3 (95% CI: 1.7–31.5; P = 0.007). However, when stratified by race, hazard ratio (HR) for discontinuation in White subjects (N = 152) was 14.4 (95% CI: 2.6–78.7; P = 0.002), but 0.8 (95% CI: 0.05–12.7; P = 0.87) for Black subjects (N = 153). The authors speculate that if jaundice as a result of hyperbilirubinemia is less obvious in individuals with darker skin, it may explain the decrease in bilirubin-associated discontinuation in Black subjects as compared to White subjects [Article:29117017]. An earlier genomewide association study (GWAS) of 475 patients administered ATV/r as part of the AIDS Clinical Trial Protocol Group A2502 previously found that the T allele at rs887829 in the promoter region of UGT1A1 (c.-364 C>T) was associated with increased peak bilirubin concentrations (P=6.4×10–12) [Article:24557078]. The T allele is in almost complete linkage disequilibrium with UGT1A1 *28, which would allow for genotyping only at rs887829, thereby facilitating the detection of the UGT1A1 *28 allele. An earlier study reported that the TT genotype at rs887829 was associated with cumulative incidence of bilirubin-related ATV/r drug-discontinuation (P = 9.3×10–14) when it was assessed in 481 Black, White, and Hispanic patients [Article:26180834].
Cholelithiasis is infrequent among HIV positive individuals treated with ATV/r, but long-term (>2 years) treatment may increase the incidence of cholelithiasis. A study of 890 Japanese patients showed a positive association between treatment duration greater than 2 years with increased likelihood of cholelithiasis, although it did not reach statistical significance in multivariate analysis [Article:24379301]. In line with these findings, a retrospective observational study of fourteen patients treated with ATV who had developed complicated cholelithiasis reported that in four patients, 100% of the biliary calculi was made up of ATV [Article:22820540]. Incidence of cholelithiasis has not been found to differ between patients treated with ATV/r and those treated with other protease inhibitors boosted with ritonavir [Article:23875004]. One retrospective pharmacogenetic study of 910 HIV-positive individuals who had undergone therapeutic drug monitoring and abdominal sonography reported no association between either UGT1A1 *28 or rs2032582 (c. c.2677 A>C/T) in ABCB1 (also known as MDR1 2677A>C/T) with risk of cholelithiasis [Article:26360703].
Nephrolithiasis is also infrequent in patients treated with ATV/r but incidence may also increase in patients treated over a year [Articles:25394067, 17502736]. Two studies reported that nephrolithiasis incidence was higher in patients treated with ATV/r as compared to other PIs (darunavir, fosamprevenir, and lopinavir) with or without ritonavir [Articles:24130871, 22820542]. A pharmacogenetic study of seventy-one Japanese patients treated with ATV/r reported that three single nucleotide polymorphisms (SNPs) in high LD within the 3’ untranslated region (UTR) of UGT1A1 were associated with nephrolithiasis: the CT genotype versus the CC genotype at rs10929303 (c.*211T>C) (adj. OR 3.7 (95% CI 1.13-11.9; P=0.030), the CG genotype versus the CC genotype at rs1042640 (c.*339G>C) (adj. OR of 5.8 (95% 1.56-21.3; P= 0.009)) and the GG or CG genotypes versus the CC genotype at rs8330 (c.*440G>C) (adj. OR of 5.8 (95% CI 1.56-21.3; P= 0.009)). Additional factors associated with increased risk of nephrolithiasis included elevated serum bilirubin concentrations and hepatitis C co-infection [Article:25151207].
Additional UGT1A Variants The structure of the UGT1A locus is complex and involves a shared set of coding exons and alternative first exons for each gene [Article:24492252]. Since the span of the whole UGT1A locus is less than 200,000 bases, variants within the boundaries upstream of the UGT1A gene may be linked to variants in UGT1A1. In a study of HIV-1 positive patients taking ATV/r (N=106) additional UGT1A variants were found to be associated with severity of hyperbilirubinemia. Patients were genotyped at rs3806596 (c.861+35196 T>C) in UGT1A3 and rs7586110 (c.855+44504 T>G) in UGT1A7, both in the respective promoter regions of those first exons. They were also genotyped at a haplotype that results in two amino acid changes in UGT1A7 (UGT1A7 129K/131K) and this haplotype is the result of several possible single nucleotide changes, which the authors do not differentiate a [Article:24492252]. rs3806596 CC, rs7586110 GG, and homozygosity of UGT1A7 129K/131K and UGT1A1 *28 was designated as the “homozygous haplotype”. Patients were sorted by the severity of hyperbilirubinemia from grade 0 (<19 µmol bilirubin/L) to grade 3-4 (= 43 µmol /L bilirubin) and the frequency of the homozygous haplotype was compared between groups as well as to a group of healthy controls. The incidence of the homozygous haplotype in healthy controls was 9.6% but in individuals with severe (grade 3 – 4) hyperbilirubinemia it was 41.2% (P< 0.0001). All six patients with grade 4 hyperbilirubinemia (severe = 85 µmol bilirubin/L) had the homozygous haplotype [Article:17058217]. When rs3806596, rs7586110 and rs17863778 were analyzed in a large genomewide association study (GWAS) of 475 patients administered ATV/r (AIDS Clinical Trial Protocol Group A2502) they were not found to be associated with increased likelihood of developing hyperbilirubinemia after correcting for presence of the rs887829 T allele in UGT1A1 [Article:24557078].
ABCB1, SLCO1B1, NR1I2 The P-gp transporter mediates the efflux of many xenobiotic substances and its function is therefore an important determinant of drug pharmacokinetics. Three polymorphisms in ABCB1 , rs1045642 (c.3435A>G), rs2032582 (c.2677A>C/T) and rs1128503 (c.1236 A>G) have been thoroughly investigated because of their effects on many drugs [Article:20216335]. Two small studies (N= 88 and N= 37) reported no association between ATV Ctrough concentrations in HIV-1 positive subjects and rs1045612 [Articles:25622064, 17324111] or rs2032582 [Article:17324111]. In 188 HIV-1 positive subjects treated with ATV/r the GG genotype at rs1045642 was associated with increased plasma concentration of ATV as compared to the AA or AG genotypes (P< 0.01) and the A allele was independently associated with lower ATV concentrations [Article:17148966]. When the three ABCB1 SNPs were assessed simultaneously (rs1128503 G, rs2032582 C, and rs1045642 G) in healthy volunteers administered ATV/r, clearance (CL/F in L/h/kg) was fastest in subjects with zero GCG copies (0.1) as compared to those with one (0.073) or two (0.070) copies of the haplotype (P < 0.03). After controlling for the number of CGC copies, race and sex were no longer associated with ATV CL/F (P = 0.86 and P = 0.21, respectively)[Article:19710077].
In a study that evaluated ATV and ritonavir concentrations in plasma and peripheral blood mononuclear cells (PBMCs) thirty-five HIV-1 positive subjects the AC and CC genotypes in rs2032582 in ABCB1 were associated with a higher ratio of ATV intracellular: plasma concentrations (P = 0.007), and significantly lower plasma ATV concentration (P= 0.043). No other SNPs were associated with ATV intracellular or plasma concentrations, although CC and CT at rs4149056 (c.521T>C) in SLCO1B1 and AG and GG at rs1523130 (c.-1663T>C) in NR1I2 were associated with increased intracellular concentrations of ritonavir [Article:24997317]. This is supported by the findings of the AIDS Clinical Trial Group A5202 GWAS which found no association between rs1045642 or rs1128503 in ABCB1, nor in rs4149056 in SLCO1B1 with ATV clearance [Article:24557078]. Altogether, little evidence suggests a potential association between the G allele at rs1045642 with increased exposure to ATV and more studies would need to confirm this. Two additional studies reported finding no significant association between polymorphisms in ABCB1 with risk of cholelithiasis or nephrolithiasis [Articles:25151207, 26360703] and a third found no association with rs2472677 (c.-22-7659C>T) in NR1I2 or rs1045642 in ABCB1 and likelihood of drug discontinuation in patients administered ATV/r [Article:21288825].
Pharmacogenomics of atazanavir without ritonavir
UGT1A1 In a single-arm, open-labelled, two-phase study of thirty-one HIV-negative volunteers, grouped by genetically determined CYP3A5 expresser status, ATV was administered alone for seven days, followed by ATV/r for another seven days and ATV pharmacokinetics and bilirubin concentrations were evaluated on days seven and fourteen. During the unboosted phase, median increases in indirect bilirubin concentrations were significantly lower in subjects with the UGT1A1 *1/*1 and UGT1A1 *1/ *28 genotypes as compared to subjects with the UGT1A1 *28/*28 genotype (P< 0.003)[Article:19710077].
In a pilot study of HIV-1 positive subjects who had been taking ATV/r as part of ART, bilirubin concentrations, immunologic, virologic and metabolic outcomes were assessed to determine whether a switch to ATV without ritonavir would be beneficial for those patients harboring UGT1A1 *28 alleles. Subjects that had one or two copies of UGT1A1 *28 were switched to the unboosted ATV regimen and had significantly lower total bilirubin when they were evaluated at twelve months (1.82 mg/dL) as compared to baseline (4.09mg/dL) (P < 0.001). In addition, these differences were accompanied by improvements in lipid profile and hepatic enzymes ( gamma-GT and transaminases) without alterations in efficacy: HIV-1 RNA was undetectable in the 24 patients that switched and there were no changes CD4 T-cell counts in any patient at 48 weeks [Article:22661571].
CYP3A5 Variability in ATV pharmacokinetics when it is administered without ritonavir has been attributed to the presence of variants in CYP3A5 that confer functionality when lack of function is more common. Individuals who carry any combination of two non-functional alleles are often designated as “non-expressers” relative to individuals with one or two functional alleles who are designated “expressers”. In the aforementioned two-phase study of thirty-one HIV-negative volunteers administered 400 mg of ATV without ritonavir for the first seven days of the study, ATV CL/F on day seven was 1.39 times faster in the “expresser” group as compared to “non-expresser” group (P=0.045). Minimum concentration (Cmin) was lower and half-life was shorter in expressers versus non-expressers but it was not significant. Significant differences were not seen in in Cmax or AUC and differences according to expresser status were only observed in the non-African-American (NAA) men and women. However, within the African-American (AA) group CL/F differences according to CYP3A5 expresser status were not observed in men (N=6) or women (N=7) although low numbers make this difficult to interpret with confidence. When controlling for presence of the GCG haplotype in ABCB1, CYP3A5 expressers still had faster CL/L versus non-expressers (P =0.01) [Article:19710077].
In a follow up to that study, fourteen of the original plasma samples were reanalyzed to determine how ATV pharmacokinetics related to the ratio of ATV-metabolites to ATV and significant differences were found when comparing between CYP3A5 expresser groups within the AA and NAA subjects. Within the NAA group, ATV AUC was 1.8 times higher in CYP3A5 non-expressers as compared to CYP3A5 expressers. ATV AUC was 2.4-fold higher in AA CYP3A5 expressers as compared to NAA CYP3A5 non-expressers. There were no significant differences between CYP3A5 expressers and non-expressers within the AA group, although there was a consistently higher M1: ATV ratio within the AA and NAA in the CYP3A5 expresser group as compared to CYP3A5 non-expresser group [Article:21148251].
In a third follow-up study, ATV pharmacokinetic parameters were re-evaluated and assessed using a population pharmacokinetic model that incorporated additional clinical and genetic variants (CYP3A4/5, ABCG2, NR1I2, and SLCO1B1). In this study, ATV CL/F was 26% slower in CYP3A5 non-expressers, 33% slower in subjects that carried one or two copies of the ABCB1 GCG haplotype, and 31% lower in subjects who carried one or more CYP3A4 *1B alleles (P=0.0074). The authors also found an interaction between CYP3A5 expresser status and genotype at rs2472677 in NR1I2. When compared to the CT and TT genotypes, the CC genotype was associated with a 37% lower CL/F in CYP3A5 non-expressers, and a 63% faster CL/F in CYP3A5 expressers. Additional demographic factors that were associated with lower CL/F were African-American ethnicity (33% decrease), female sex (25% decreased), and age (1.7% decrease/year over age 30)[Article:22394315].
A subset of the retrospective Prospective Evaluation of Antivirals in a Resource Limited Settings study (PEARLS) included sixty-nine treatment-naïve HIV-positive subjects treated with ATV without ritonavir from the United States, Peru and South Africa. A single plasma sample was collected at four and eight weeks and population pharmacokinetic modeling was used to predict CL/F and C24, metabolite to parent compound ratios (M1: ATV and M2: ATV) as well as likelihood of virologic failure by CYP3A5 expresser status, or genotype. CYP3A5 expressers had a statistically significant decrease in likelihood of virologic failure (OR 0.95 (95% CI 0.89-1) as compared to CYP3A5 non-expressers (OR 0.73 (95% CI 0.57-0.94) P=0.026) at 72 weeks. No statistically significant associations were found between ATV CL/F, C24, M1:ATV or M2:ATV with CYP3A5 expresser status after adjusting for race and gender, or in ranked analysis. When subjects were grouped by genotype rather than by CYP3A5 status, ATV CL/F was found to be significantly faster for subjects with the CYP3A5 *1/*1 as compared to CYP3A5 *1/*3 and CYP3A5 *3/ *3 (P=0.0006) genotypes and the difference with *1/ *3 when adjusting for race and gender (P= 0.001). Surprisingly, the CYP3A5 *1/ *6 genotype was associated with faster CL/F (15.7 (14.6-16.8)) as compared to CYP3A5 *1/ *1 genotype (12.4 ng/ml) (11.6-13.4)) (P= 0.004) even after adjusting for race and gender. CYP3A5 genotypes were not associated with C24, nor M1: ATV or M2: ATV after adjusting for race and gender or in ranked analysis [Article:26892777].
ABCB1, SLCO1B1, NR1I2 In a two-part study, in which healthy volunteers were administered ATV followed by ATV/r, several pharmacokinetic parameters such as CL/F and Cmin of ATV were determined. During the phase in which ATV was administered without ritonavir, ATV CL/F was reported to be faster, and Cmin was reported to be lower, respectively, in subjects with zero versus one or two copies of the GCG haplotype in ABCB1 (P < 0.007 for CL/F and P < 0.02 for Cmin). Homozygosity for the GCG haplotype in ABCB1 was also associated with higher bilirubin concentration as compared to zero copies (P = 0.036). [Article:19710077]. In a separate study of seventy-four White patients administered ATV only, only the G allele at rs1045642 in ABCB1 was found to be associated with higher median plasma concentrations of ATV and median ATV Cmin was also higher in patients with the GG genotype as compared to the AA and AG genotypes (P < 0.01). A greater proportion of subjects with the GG genotype (92%) also had bilirubin concentrations above the upper limit of normal (ULM > 1.3 mg/dl), as compared to patients with the AG (67%) (P<0.05) or AA genotypes (41 %) (P<0.05) [Article:16355344].
Polymorphisms in NR1I2, SLCO1B1 and ABCB1 were also evaluated in PEARLS study. That study included people from Peru, South Africa and the U.S.A (N = 69) of various ethnicities who were co administered unboosted ATV with didanosine-EC + emtricitabine. In that study, the CT genotype at rs2472677 in NR1I2 was associated with a significant decrease in ATV C24 (48.4 ng/ml) as compared to either the CC (126.8 ng/ml) or TT genotypes (172.6 ng/ml) (P = 0.01 and P = 0.007, respectively) even after adjustment for race and sex. Subjects with two copies of ABCB1 GCG haplotype had decreased ATV C24 as compared to subjects with one, although the association did not remain significant after adjusting for race and gender or in ranked analysis. Neither rs2306283, nor rs4149056 in SLCO1B1 were associated with CL/F, C24, M1: ATV or M2: ATV [Article:26892777]. This is contradicted by two other studies in which HIV-1 positive patients were administered ATV without ritonavir. In the first study (N = 109; 26% female, ethnicities unknown) the TT genotype at rs2472677 in NR1I2 was associated with lower median trough concentrations of ATV (Ctrough) (34 ng/mL versus 152 ng/mL; P = .001), and an increased likelihood that ATV concentrations were below the minimum effective concentration of 150 ng/mL in two cohorts (OR 18 95% CI (2.1 -153.9) P = 0.008). In addition, tenofovir was not associated with Ctrough in either group [Article:18831695]. In the second study, which used many subjects from the same cohort as well as a new cohort (N = 182; 64% male), population pharmacokinetic modeling was used to show that the CT and CC genotypes at rs2472677 in NR1I2 were associated with a 17.2% increase in CL/F as compared to the TT genotype [Article:20921307].
Finally, a randomized controlled pilot study investigated whether pharmacogenetics-based dosing could improve ATVs pharmacokinetic profile in patients switched to 400 mg ATV without ritonavir due to issues of toxicity, tolerability or dose simplification. The pharmacogenetics-guided dosing arm (N = 40) was assigned to a dosing schedule guided by genotypes at three different SNPs: rs2472677 in NR1I2, rs1045642 in ABCB1, and rs4149056 in SLCO1B1. The TT genotype at rs2472677, AA or AG genotypes at rs1045642, and the TT genotype at rs4149056 were considered “unfavorable genotypes” because they had previously been associated with decreased exposure to ATV. Each unfavorable genotype was given one point and patients with two to three points received twice daily 200 mg doses of ATV to improve ATV exposure whereas those with zero or one point received 400 mg ATV once daily. Patients in the non-pharmacogenomics-guided dosing arm (N=40) received a once daily dose of 400 mg of ATV without regard to genotype. The geometric mean of ATV Ctrough in the pharmacogenetics arm was values was 253 ng/mL (150–542) versus 111 ng/mL (64–190) in the standard-dose arm. In addition, 75.7% of the subjects in the pharmacogenetics arm had ATV Ctrough concentrations within the therapeutic range (>150 ng/ml) while only 38.9% of subjects in the standard dose arm did (P = 0.001; RR 4.89, 95% CI 1.79–13.38)[Article:26174719]. Current evidence, along with the pharmacogenetics-guided dosing study, may support the association between the TT genotype at rs2472677 in NR1I2 and the G allele at rs1045642 in ABCB1 with increased ATV exposure when it is administered without a pharmacokinetic enhancer.
Conclusions
The findings summarized here are limited by the small cohort sizes and the overrepresentation of White, male subjects in most studies thereby minimizing the extent to which they may be extrapolated to more diverse populations. Future studies in larger and more diverse cohorts could help to explain why CYP3A5 appears to affect unboosted ATV differently in Black versus White subjects. In addition, the incorporation of genetic variants known to affect ritonavir pharmacokinetics into a dosing algorithm that includes genetic variants known to affect ATV pharmacokinetic may help to more precisely estimate dosing strategies for individual patients, although dose reductions in ATV and/or ritonavir have also shown promise in some patients [Article:23011396]. There is also good and consistent evidence of an association is between UGT1A1 rs8175347 (*28, *37) and the TT genotype at rs8175347 with increased likelihood of hyperbilirubinemia in patients administered ATV/r, as well as an increased likelihood of drug-discontinuation. The Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for pharmacogenomic prescribing of ATV, recommends discussing the high likelihood of developing jaundice in patients homozygous for these alleles as well as considering alternative ART regimens unless jaundice is not of concern to the patient [Article:26417955]. Other variants in other genes may influence severity of hyperbilirubinemia or nephrolithiasis but have not been replicated and are not yet clinically actionable. An effective pharmacogenetics-guided dosing strategy will likely require larger studies, in more diverse cohorts in order to uncover additional genetic and non-genetic factors, such as race, sex, concomitant drug use or co-infection to improve drug adherence and minimize adverse events in patients receiving ATV.
Reactions & interactions (18)
-
Biochemical Reaction
atazanavir → atazanavir M2
-
Biochemical Reaction
atazanavir → atazanavir M4
-
Biochemical Reaction
atazanavir → atazanavir M1
-
Biochemical Reaction
atazanavir → atazanavir M5
-
Biochemical Reaction
atazanavir → atazanavir M3
-
Catalysis
CYP3A4 → Biochemical Reaction
-
Catalysis
CYP3A5 → Biochemical Reaction
-
Catalysis
CYP3A5 → Biochemical Reaction
-
Catalysis
CYP3A4 → Biochemical Reaction
-
Catalysis
CYP3A5 → Biochemical Reaction
-
Catalysis
CYP3A4 → Biochemical Reaction
-
Catalysis
CYP3A4 → Biochemical Reaction
-
Catalysis
CYP3A5 → Biochemical Reaction
-
Catalysis
CYP3A5 → Biochemical Reaction
-
Catalysis
CYP3A4 → Biochemical Reaction
-
Inhibition
atazanavir → UGT1A1
-
Transport
atazanavir → atazanavir
-
Transport
atazanavir → atazanavir
Edit history (4)
- 2017-10-09 Create
- 2018-07-10 Update Updated citation information.
- 2024-07-30 Update fixed typos
- 2025-07-17 Update Removed broken links